Controlling growth of self-propagating molecular assemblies†
Leila
Motiei
a,
Moran
Feller
a,
Guennadi
Evmenenko
b,
Pulak
Dutta
b and
Milko E.
van der Boom
*a
aDepartment of Organic Chemistry, The Weizmann Institute of Science, Rehovot, Israel. E-mail: milko.vanderboom@weizmann.ac.il; Fax: +972 8934 4142; Tel: +972 8934 2515
bDepartment of Physics and Astronomy, Northwestern University, Evanston, IL 60208-3113, USA
First published on 23rd September 2011
Abstract
A series of six self-propagating molecular assemblies (SPMAs) were generated by alternative solution-deposition of ruthenium polypyridyl complexes and d8palladium and platinum salts on glass and silicon substrates. The d6 polypyridyl complexes have three pyridine units available for forming networks by coordination to the metal salts. This two-step film growth process is fast (15 min/step) and can be carried out conveniently under ambient conditions in air. The reactivity of the common metal salts (ML2X2: M = Pd, X = Cl, L = PhCN, ½ 1,5-cyclooctadiene (COD), SMe2 and M = Pt, X = Cl, Br, I, L = PhCN) is a dominant factor in the film growth. Although the assembly structures are comparable, their exponential growth can be controlled by varying the metals salts. The co-ligands, halides, and metal centers can be used to control the film thicknesses and light absorption intensities of the metal-to-ligand charge transfer (MLCT) bands by a factor of ∼3.5 for 13 deposition steps, whereas the surface morphologies and molecular densities of the SPMAs are similar. The surface-confined assemblies have been characterized using a combination of optical (UV/Vis, ellipsometry) spectroscopy, atomic force microscopy (AFM), X-ray photoelectron spectroscopy (XPS), and synchrotron X-ray reflectivity (XRR).
Introduction
The formation of coordination metal–organic assemblies is an intriguing but often complex process that involves many variables. Numerous fascinating structures have been reported in solution and as solid-state materials with possible applications ranging from sensing and catalysis to energy storage.1–4 For example, Nitschke et al. reported the stabilization of white phosphorus within a self-assembled tetrahedral capsule.4 The design of such materials with precise control over their structure and properties is a difficult task. To understand the underlying formation mechanism for a given metal–organic assembly and to be able to predict structures and function with a high level of precision, one must vary the experimental reaction parameters systematically. Such an experimental approach allows one to identify those factors that play dominant roles and to possibly determine the synergy between them.Several methods have been developed to form metal–organic solids and films, including the stepwise solution-based deposition of ligands and metal ions on various substrates.5–8 The formation of such coordination-based multilayers has been studied in much detail for more than two decades and has resulted in diverse materials. Alternating solution-based deposition has also afforded metal–organic frameworks (MOFs) with unique structural features not attainable by other routes.1,9 We have shown that chromophores 1–6 (Scheme 1) and a palladium salt can be used for the stepwise formation of solid-state metal–organic structures, including 3D-ordered assemblies.10,11 Metal-mediated layer-by-layer (LbL) growth of all-organic films is relatively rare.12
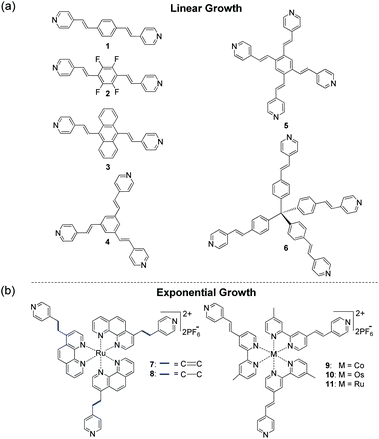 | ||
| Scheme 1 Molecular structures for the stepwise generation of coordination-based films with PdCl2(PhCN)2. The use of compounds 1–6 resulted in linear growth (a),10,11 whereas self-propagating molecular assemblies (SPMAs) were obtained with complexes 7–11 (b).13–17 | ||
Regardless of the wide variety of metal ions and organic ligands used, the aforementioned surface-confined assemblies are formed by linear processes.5,6,9–11 Physicochemical properties, including material thickness and light absorption intensities, are linearly correlated with the number of deposition steps. The surfaces of these linearly growing metal–organic assemblies are efficient binding sites for incoming metal ions or ligands from solution, and they can direct the supramolecular structure of the incoming layer. The bulk of the assembly, however, does not play a significant role during the growth process.
Exponentially growing assemblies are intriguing because the material being formed is involved in its own formation. A rare example is Sagiv et al.'s self-replicating organosilane-multilayers.18Polyelectrolyte-based films may also exhibit nonlinear growth.19–22 We have shown that the alternative deposition of polypyridyl ruthenium, osmium, and cobalt complexes (Scheme 1; 7–11) as well as a d8palladium salt, PdCl2(PhCN)2, from solution results in exponentially growing metal–organic assemblies.13–17 The overall process is a three-component process in which the self-propagating molecular assemblies (SPMAs) store palladium. This excess of palladium is used to bind the polypyridyl complexes to the surface. This d8 metal is an important structural synthon for forming metal–organic networks. Pyridine and derivatives thereof readily coordinate to PdCl2 in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ligand-to-metal ratio forming stable 16-electron complexes having a square-planar geometry around the metal center.10 However, the role of the metal complex precursor in SPMA formation has not been evaluated. Until now, all SPMAs have been generated from Pd(PhCN)2Cl2.13–17 We have shown that the growth of the SPMAs can be systematically controlled by changing the structure of the molecular component (Scheme 1) and by varying the reaction conditions. Linear growth is observed with polypyridyl complexes (8) when the surface-bound assemblies are only briefly exposed to the palladium salt.14 Combining organic chromophores (4) with polypyridyl complexes (11)14 in one assembly results in enhanced exponential growth.13 The redox-active SPMAs have been used (i) to study electron-transfer and electrochromism,16 (ii) to fabricate inverted solar cells,15 and (iii) to demonstrate Boolean and sequential logic operations.23
:1 ligand-to-metal ratio forming stable 16-electron complexes having a square-planar geometry around the metal center.10 However, the role of the metal complex precursor in SPMA formation has not been evaluated. Until now, all SPMAs have been generated from Pd(PhCN)2Cl2.13–17 We have shown that the growth of the SPMAs can be systematically controlled by changing the structure of the molecular component (Scheme 1) and by varying the reaction conditions. Linear growth is observed with polypyridyl complexes (8) when the surface-bound assemblies are only briefly exposed to the palladium salt.14 Combining organic chromophores (4) with polypyridyl complexes (11)14 in one assembly results in enhanced exponential growth.13 The redox-active SPMAs have been used (i) to study electron-transfer and electrochromism,16 (ii) to fabricate inverted solar cells,15 and (iii) to demonstrate Boolean and sequential logic operations.23
In the present study we show that the choice of the metal-ion source significantly affects the formation, optical properties, and thickness of the SPMAs. Six SPMAs were generated on silicon and glass substrates with a ruthenium polypyridyl complex (11)14,15 and a series of metal salts of the formula: ML2X2 with M = Pd, X = Cl, L = PhCN, ½ 1,5-cyclooctadiene (COD), SMe2 and M = Pt, X = Cl, Br, I, L = PhCN). These transition metal ions, halides, and co-ligands play not only a key structural role by coordination to the polypyridyl complexes to generate the surface-confined networks but are also important for efficient film assembly. The reactivity of the metal salts is expressed in the SPMAs' properties. We demonstrate here that both absorption intensities and thicknesses of the SPMAs can be varied by a factor of ∼3.5, while a similar surface morphology and molecular density is maintained.
Results and discussion
p-Chlorobenzyl-based monolayers24 covalently attached to glass and silicon substrates were used to generate template layers (TLs) consisting of the pyridinium salt of complex 11 (Scheme 2a).15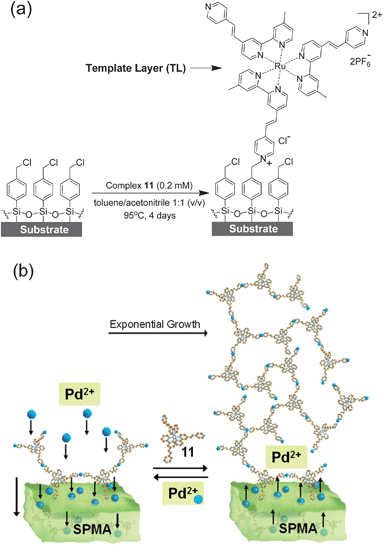 | ||
| Scheme 2 (a) Glass and silicon substrates functionalized with a p-chlorobenzyl-terminated monolayer24 were reacted at elevated temperatures with a dry acetonitrile/toluene solution of complex 11 for 4 days under an inert atmosphere with the exclusion of light to generate a template layer (TL = deposition step 1).14,15X-ray photoelectron spectroscopy (XPS) studies have shown that the analogous complexes 7 and 10 (Scheme 1) are only bound via one pyridine moiety.14,17 (b) Schematic presentation of the formation of SPMAs with complex 11 and PdCl2(PhCN)2.13–17 | ||
Subsequently, the six SPMAs were formed by iterative immersion of the functionalized substrates for 15 min into a THF solution of one of the Pd or Pt precursors and a THF/DMF (9:1, v/v) solution of complex 11. The SPMAs were thoroughly washed and sonicated in organic solvents between each deposition step (for details, see Materials and methods).
The SPMAs were characterized by UV/Vis spectrometry, atomic force microscopy (AFM), ellipsometry, X-ray photoelectron spectroscopy (XPS) and synchrotron X-ray reflectivity (XRR) measurements. The optical absorption intensities of the metal-to-ligand charge transfer (MLCT) bands at λ = 495 nm and the thickness of the SPMAs versus the number of deposition steps show that exponential growth occurs (Fig. 1, 3 and 4). The half-width and the maximum position of the absorption bands are not affected during the growth process, as shown for SPMA|Pd(PhCN)2Cl2 in Fig. 1, suggesting no strong inter- and intramolecular interactions. Semicontact AFM measurements show that the SPMAs have comparable film surfaces after 13 deposition steps with a low root-mean-square surface roughness (Rrms) of 0.4–1.1 nm for scan areas of 0.5 nm × 0.5 nm, with no apparent grain boundaries (Fig. 2 and Fig. S2†). These surface morphologies are common for related molecule-based multilayers that exhibit linear growthversus the number of deposition steps.10,11 The optical and AFM data of all SPMAs are shown in Fig. S1 and S2, respectively.†
 | ||
| Fig. 1 Formation of SPMA|Pd(PhCN)2Cl2: Representative UV/Vis absorption spectra on glass substrates. These ex-situspectra were recorded after exposing the SPMA to a THF/DMF (9:1, v/v) solution containing complex 11 (0.2 mM) for 15 min. The red spectrum is from the template layer (TL). The baseline is shown in black. | ||
 | ||
| Fig. 2 Representative semicontact AFM image of SPMA|Pd(PhCN)2Cl2 after 13 deposition steps of complex 11 on silicon substrates (Scheme 2). Step 1 is the template layer (TL). A scan area of 500 nm × 500 nm is shown with a roughness, Rrms, of 0.4 nm. UV/Vis spectra and AFM images of all the SPMAs are available in the ESI (Fig. S1 and S2).† | ||
The growth of the SPMAs is clearly affected by the nature of the metal complex precursor, as can been seen by plotting the absorption intensities and film thickness versus the number of deposition steps (Fig. 3 and 4). Steep exponential growth is achieved with Pd(PhCN)2Cl2, which is in contrast with the build-up observed with Pt(PhCN)2Cl2 (Fig. 3). Thus, varying the metal ion of the metal complex precursors results in significant absorption intensity enhancement (3.5×) and a difference of 11 nm in the final SPMA thickness (Table 1, entries 1 and 4). Pd(PhCN)2Cl2 is the more reactive precursor. The halide also plays an important role. The exponential growth increases in the order: Pt(PhCN)2I2 > Pt(PhCN)2Br2 ≥ Pt(PhCN)2Cl2 (Fig. 3; Table 1, entries 1–3). Whereas the difference between the Br and Cl derivatives is minor, the use of Pt(PhCN)2I2 resulted in exponential growth similar to the one observed with Pd(PhCN)2Cl2. The ‘bulkiness’ of halogen atoms is known to accelerate ligand exchange process.25 Our observations might suggest that the reactivity of the metal complex precursors plays a dominant role.26–28 This hypothesis is fully supported by the SPMA growth upon varying the organic ligands (PdL2Cl2; L = PhCN, ½ COD and SMe2). The SPMA growth follows the following order: Pd(PhCN)2Cl2 > Pd(COD)Cl2 > Pd(SMe2)2Cl2 (Fig. 4). Also here the differences are significant. The growth is enhanced by ∼2.8× for 13 deposition steps by varying these co-ligands (Table 1, entries 4–6). The SPMA growth is directed related to the relative reactivity/stability of these three palladium salts. Pd(PhCN)2Cl2 reacts readily with COD in solution to form Pd(COD)Cl2, which is more stable by 13 kcal mol−1.28 In addition, Pd(COD)Cl2 and Pd(PhCN)2Cl226 react with two equivalents of SMe2 to form Pd(SMe2)2Cl2.
 | ||
Fig. 3 Exponential dependence of (a) the intensities of the metal–ligand charge transfer (MLCT) bands at λ ≈ 495 nm on glass substrates, and (b) the film thicknesses on silicon for (■) SPMA|Pt(PhCN)2Cl2, ( ) SPMA|Pt(PhCN)2Br2, ( ) SPMA|Pt(PhCN)2Br2, ( ) SPMA|Pt(PhCN)2I2, and ( ) SPMA|Pt(PhCN)2I2, and ( ) SPMA|Pd(PhCN)2Cl2versus the number of deposition steps (R2 = 0.99). Step 1 is the template layer (TL). The film thicknesses were obtained by spectroscopic ellipsometry. ) SPMA|Pd(PhCN)2Cl2versus the number of deposition steps (R2 = 0.99). Step 1 is the template layer (TL). The film thicknesses were obtained by spectroscopic ellipsometry. | ||
 | ||
Fig. 4 Exponential dependence of (a) the intensities of the metal–ligand charge transfer (MLCT) bands at λ ≈ 495 nm, and (b) the film thicknesses on silicon for (■) SPMA|Pd(SMe2)2Cl2, ( ) SPMA|Pd(COD)Cl2, and ( ) SPMA|Pd(COD)Cl2, and ( ) SPMA|Pd(PhCN)2Cl2versus the number of deposition steps (R2 = 0.99). Step 1 is the template layer (TL). The film thicknesses were obtained by spectroscopic ellipsometry. ) SPMA|Pd(PhCN)2Cl2versus the number of deposition steps (R2 = 0.99). Step 1 is the template layer (TL). The film thicknesses were obtained by spectroscopic ellipsometry. | ||
| Entry | M | L | X | T/nm | A a | E b | M/Nc | M/Ruc | N/Ru |
|---|---|---|---|---|---|---|---|---|---|
| a Absorption at λ ≈ 495 nm ×10−2. b E = Enhancement of the growth derived from the thickness (T) and the optical data (A) (normalized to entry 1). c M = Pt or Pd; the ratios were obtained by XPS. d The S/Pd ratio is 0.28. The XPS spectra were recorded at take-off angles of θ = 0° and 45.0° and show similar elemental ratios. | |||||||||
| 1 | Pt | PhCN | Cl | 5.5 | 3.9 | 1.0 | 0.20 | 2.7 | 13 |
| 2 | Pt | PhCN | Br | 6.6 | 4.7 | 1.2 | 0.17 | 1.8 | 11 |
| 3 | Pt | PhCN | I | 17.0 | 12 | 3.1 | 0.19 | 1.9 | 10 |
| 4 | Pd | PhCN | Cl | 18.1 | 15 | 3.6 | 0.26 | 2.8 | 11 |
| 5 | Pd | ½COD | Cl | 11.2 | 9.2 | 2.2 | 0.16 | 1.5 | 9 |
| 6 | Pd | SMe2d | Cl | 6.4 | 5.8 | 1.3 | 0.14 | 1.5 | 11 |
The structural regularity of the SPMAs is evident from the good linear dependence of the absorption intensities versus the film thicknesses (Fig. 5, Table S1†; R2 ≥ 0.98), indicating that the molecular density and the relative roughness remain nearly constant during the growth process. The average density of complex 11 for the fully formed SPMAs is roughly estimated from the optical and thickness data to be 1.1 (Pt) and 1.3 (Pd) molecules nm−3 and is very similar for each deposition step of complex 11 (Table S1†). Such high values have been reported for single-crystal packing densities of polypyridyl complexes.29 Moreover, the similarity between the slopes of the Pd (8.1–10 × 10−4) and the Pt (6.4–7.1 × 10−4) SPMAs indicate that the structures are comparable (Table S1†), but are affected by the different metal. The palladium salts might seem to form slightly more densely packed SPMAs.
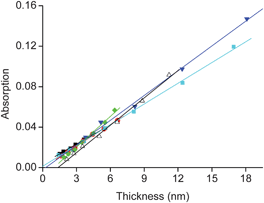 | ||
Fig. 5 The SPMA absorption intensity at λ ≈ 495 nm (Fig. 3a and 4a) versus thickness (Fig. 3b and 4b). The 6 lines are linear fits with R2 ≥ 0.98 for (■) SPMA|Pt(PhCN)2Cl2, ( ) SPMA|Pt(PhCN)2Br2, ( ) SPMA|Pt(PhCN)2Br2, ( ) SPMA|Pt(PhCN)2I2, ( ) SPMA|Pt(PhCN)2I2, ( ) SPMA|Pd(PhCN)2Cl2, ( ) SPMA|Pd(PhCN)2Cl2, ( ) SPMA|Pd(SMe2)2Cl2, and (△) SPMA|Pd(COD)Cl2. The slopes are listed in Table S1.† ) SPMA|Pd(SMe2)2Cl2, and (△) SPMA|Pd(COD)Cl2. The slopes are listed in Table S1.† | ||
The SPMA formation was further confirmed by XRR measurements of SPMA|Pt(PhCN)2Br2 and SPMA|Pd(COD)Cl2 after 13 deposition steps (Fig. 6 and S3†). We observed film thicknesses of 6 and 1.1 nm, respectively, with a relative roughness of ∼20%. These thickness values are in agreement with the ellipsometry data (Table 1, entries 2 and 5). The electron density, ρ, for the SPMA|Pd(COD)Cl2 is ∼11% higher (Fig. 6b), which is in agreement with the above conclusions. We have previously shown by XRR measurements that ρ is constant during the build-up of a SPMA formed with osmium complex 10 and Pd(PhCN)2Cl2 (Scheme 1).17
 | ||
| Fig. 6 (a) Representative synchrotron X-ray reflectivity (XRR) spectrum of SPMA|Pt(PhCN)2Br2 on a silicon substrate (see Fig. S3† for the spectrum of SPMA|Pd(COD)Cl2). The blue trace is a fit of the experimental data (black dots).30 (b) Electron density plots for SPMA|Pt(PhCN)2Br2 (blue line) and SPMA|Pd(COD)Cl2 (red line) after 13 deposition steps with step 1 being the template layer (TL). | ||
The XPS-derived elemental ratios are centred around the theoretical values expected for a fully formed network where two pyridinium groups are coordinated to a palladium or platinum centre: M/N = 0.17; M/Ru = 1.5 and N/Ru = 9 (M = Pt or Pd). No clear trend is apparent (Table 1; entries 1–6). Angle-dependent XPS measurements indicate the formation of homogeneous structures. Some of the SPMAs contain some of the d8 metal salts and/or free ligands. For instance, SPMA|Pd(SMe2)2Cl2 contains sulfur as a minor component (S/Pd = 0.28).
Summary and conclusions
Regardless of the metal salt used, all SPMAs form regularly growing assemblies on glass and silicon substrates. Interestingly, the different metal salts do not seem to drastically affect the surface morphology of the SPMAs, the elemental ratios, molecular density, and the half-width as well as the maximum position of the absorption bands. The nature of the metal complex precursors significantly affects the SPMA growth, but our observations indicate that the overall structure of the SPMAs is similar. The following parameters were noted for promoting efficient film growth: (i) Pd > Pt, (ii) I > Br ≥ Cl, and (iii) PhCN > COD > SMe2. These trends can be explained by the reactivity of these metal salts with the surface-bound polypyridyl complexes.26,27 Ligand substitution reactions with Pd tend to be faster in comparison with the corresponding Pt complexes. The metal–ligand bond strength of the precursors also affects the growth. A higher reactivity results in more efficient growth because more Pd or Pt is accessible for the system. The different dimensions of the metal salts studied here seem to play a minor role. These findings are in good agreement with our mechanistic interpretation of the film growth where the SPMAs are reservoirs of metal salts.14,17 This model is to some extent similar to the one operating with exponentially growing polyelectrolyte-based materials.19,31 However, the film roughness might also play a role. This issue is currently under debate.32 At present, we cannot exclude that the surface morphology affects the SPMA growth. Fine tuning of the SPMAs' composition without compromising the efficiency of the film growth can be achieved by combining parameters having opposite effects. For instance, much more efficient growth was observed with Pd(PhCN)2Cl2 than with Pt(PhCN)2Cl2. However, Pd(PhCN)2Cl2 and Pt(PhCN)2I2 are equally efficient. In order to grow SPMAs with different metals (Pd or Pt) having similar properties (i.e., light absorption, thickness, and roughness), one has to use different halides as well. These parameters (i.e., Pt, I) may affect the molecular density, which might be interesting for size-selective sensing. A few other studies have addressed the use of different metal complex precursors with linear growing metal–organic multilayers and oligomers.6,8 Of course, there remain many other parameters to be explored, to gain a better and more thorough picture of the exponential growth. Solvents,33 temperature,20 and the anions21 of the polypyridyl complexes might also affect the growth and the SPMA structures. However, the metal salts are certainly of prime importance for the exponential growth and they can be used to control it.Materials and methods
PtCl2, PtBr2, and PtI2 were purchased from Aldrich and used as received. Solvents (AR grade) were purchased from Bio-Lab (Jerusalem) and Aldrich. trans-Pt(PhCN)2X2, (X = Cl, Br, I),34trans-Pd(PhCN)2Cl2,35Pd(COD)Cl2,36trans-Pd(SMe2)2Cl2,37 and complex 11 were prepared according to procedures in the literature.14 Single-crystal silicon (100) substrates were purchased from Wafernet (San Jose, CA) and were cleaned by sonication in hexane followed by acetone, then ethanol, and dried under an N2 stream. Subsequently, they were cleaned for 20 min with UV and ozone in a UVOCS cleaning system (Montgomery, PA). Float glass (Corning, NY) was cleaned by immersion in a “piranha” solution (7:3 (v/v) H2SO4/30% H2O2) for 1 h. Caution: piranha solution is an extremely dangerous oxidizing agent and should be handled with care using appropriate personal protection. Subsequently, the substrates were rinsed with deionized (DI) water followed by the RCA cleaning protocol: 1:5:1 (v/v) NH4OH/H2O/30% H2O2 at room temperature for 45 min. The substrates were washed with ample amounts of DI water and were dried under an N2 stream. All substrates were then dried in an oven for 2 h at 130 °C. UV/Vis spectra were recorded on a Cary 100 spectrophotometer. Spectroscopic ellipsometry was recorded on an M 2000V (J. A. Wollam Co., Inc.) instrument with VASE32 software. AFM images were recorded using a Solver P47 (NT-MDT, Russia) operated in semicontact mode. All measurements were carried out at 295 K unless otherwise mentioned. X-ray reflectivity (XRR) measurements were performed at beamline X18A of the synchrotron National Synchrotron Light Source, Brookhaven (USA) using a four-circle Huber diffractometer in the specular reflection mode (i.e., incident angle was equal to the exit angle). An X-ray beam with an energy of E = 10.0 keV (λ = 1.240 Å) was used. The beam size was 0.50 mm vertically and 1.0 mm horizontally. The samples were held under a helium atmosphere during the measurements to reduce radiation damage and the background scattering from the ambient gas. The off-specular background was measured and subtracted from the specular counts. X-ray photoelectron spectroscopy (XPS) measurements were carried out on glass substrates with a Kratos AXIS ULTRA system using a monochromatized Al Kα X-ray source (hν = 1486.6 eV) at 75 W and detection pass energies ranging between 10 and 80 eV. Angular resolved spectra were recorded at θ = 0° and 45.0°, where θ is the take-off angle with respect to the surface normal. A low energy flood gun was used to neutralize charging while measuring glass substrates.
Template layer (TL) formation
Glass and silicon substrates (0.8 cm × 2.5 cm) functionalized with a p-chlorobenzyl-terminated monolayer24 were loaded into a glass pressure vessel and immersed in a dry acetonitrile/toluene (1:1 v/v) solution of complex 1114 (0.2 mM) and heated for 96 h at 95 °C with the exclusion of light (Scheme 2a). The TLs were then rinsed with acetonitrile and sonicated for 5 min each in acetonitrile, acetone and isopropanol. The samples were then carefully wiped with a task wipe and dried under a stream of N2. The TL is deposition step 1 of the SPMA formation.
SPMA formation
Glass and silicon substrates (0.8 cm × 2.5 cm) functionalized with the 11-based TLs (= deposition step 1; Scheme 2a) were loaded onto a Teflon holder and immersed for 15 min in a 1 mM solution of a metal salt (ML2X2: M = Pd, X = Cl, L = PhCN, ½ 1,5-cyclooctadiene (COD), SMe2 and M = Pt, X = Cl, Br, I, L = PhCN) in THF. The samples were then sonicated twice in THF and once in acetone for 2 min each. Subsequently, the samples were immersed for 15 min in a 0.2 mM solution of compound 11 in THF/DMF (9:1, v/v). The samples were then sonicated twice in THF and once in acetone for 2 min each. This procedure was repeated 6 times (=12 deposition steps). Finally, the samples were rinsed in ethanol and dried under a stream of N2. This procedure was carried out in air at room temperature.
Acknowledgements
This research was supported by the Helen and Martin Kimmel Center for Molecular Design, the Israel Science Foundation (289/09), MINERVA, and by the U.S. National Science Foundation under Grant No. DMR-1006432. The X-ray reflectivity measurements were performed at Beamline X18A of the National Synchrotron Light Source, supported by the U.S. Department of Energy under contract no. DE-AC02-98CH10886. X-ray photoelectron spectroscopy (XPS) measurements were performed by Drs T. Bendikov and H. Cohen (Department of Chemical Research Support, The Weizmann Institute of Science).Notes and references
- D. Zacher, R. Schmid, C. Wöll and R. A. Fischer, Angew. Chem., Int. Ed., 2011, 50, 176 CrossRef CAS.
- A. Carne, C. Carbonell, I. Imaz and D. Maspoch, Chem. Soc. Rev., 2011, 40, 291 RSC; R. Mas-Ballesté, J. Gómez-Herrero and F. Zamora, Chem. Soc. Rev., 2010, 39, 4220 RSC; L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC; B. H. Northrop, Y.-R. Zheng, K.-W. Chi and P. J. Stang, Acc. Chem. Res., 2009, 42, 1554 CrossRef CAS; C. G. Oliveri, P. A. Ulmann, M. J. Wiester and C. A. Mirkin, Acc. Chem. Res., 2008, 41, 1618 CrossRef.
- X. de Hatten, N. Bell, N. Yufa, G. Christmann and J. R. Nitschke, J. Am. Chem. Soc., 2011, 133, 3158 CrossRef CAS; R. Kaminker, R. Popovitz-Biro and M. E. van der Boom, Angew. Chem., Int. Ed., 2011, 50, 3224 CrossRef; M. Marschall, J. Reichert, A. Weber-Bargioni, K. Seufert, W. Auwarter, S. Klyatskaya, G. Zoppellaro, M. Ruben and J. V. Barth, Nat. Chem., 2010, 2, 131 CrossRef; S. Gao, Z. Zheng, L. Jian and R. Cao, Chem. Commun., 2010, 46, 7584 RSC; O. K. Farha, C. D. Malliakas, M. G. Kanatzidis and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 950 CrossRef; H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846 CrossRef; G. Golubkov, H. Weissman, E. Shirman, S. G. Wolf, I. Pinkas and B. Rybtchinski, Angew. Chem., Int. Ed., 2009, 48, 926 CrossRef.
- P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697 CrossRef CAS.
- K. Kanaizuka, S. Kato, H. Moriyama and C. Pac, J. Mater. Res., 2011, 26, 236 CrossRef CAS; W. Zhao, B. Tong, J. Shi, Y. Pan, J. Shen, J. Zhi, W. K. Chan and Y. Dong, Langmuir, 2010, 26, 16084 CrossRef; H. E. Katz, G. Scheller, T. M. Putvinski, M. L. SchiIlling, W. L. Wilson and C. E. D. Chidsey, Science, 1991, 254, 1485 Search PubMed.
- A. Hatzor, T. Moav, H. Cohen, S. Matlis, J. Libman, A. Vaskevich, A. Shanzer and I. Rubinstein, J. Am. Chem. Soc., 1998, 120, 13469 CrossRef CAS; I. Rubinstein and A. Vaskevich, Isr. J. Chem., 2010, 50, 333 CrossRef; G. Cao, H. G. Hong and T. E. Mallouk, Acc. Chem. Res., 1992, 25, 420 CrossRef.
- N. Tuccitto, I. Delfanti, V. Torrisi, F. Scandola, C. Chiorboli, V. Stepanenko, F. Würthner and A. Licciardello, Phys. Chem. Chem. Phys., 2009, 11, 4033 RSC.
- N. Tuccitto, V. Ferri, M. Cavazzini, S. Quici, G. Zhavnerko, A. Licciardello and M. A. Rampi, Nat. Mater., 2009, 8, 41 CrossRef CAS; T. Kurita, Y. Nishimori, F. Toshimitsu, S. Muratsugu, S. Kume and H. Nishihara, J. Am. Chem. Soc., 2010, 132, 4524 CrossRef; Y. Liang and R. H. Schmehl, J. Chem. Soc., Chem. Commun., 1995, 1007 RSC.
- R. Makiura, S. Motoyama, Y. Umemura, H. Yamanaka, O. Sakata and H. Kitagawa, Nat. Mater., 2010, 9, 565 CrossRef CAS; O. Shekhah, H. Wang, M. Paradinas, C. Ocal, B. Schupbach, A. Terfort, D. Zacher, R. A. Fischer and C. Wöll, Nat. Mater., 2009, 8, 481 CrossRef.
- R. Kaminker, L. Motiei, A. Gulino, I. Fragalà, L. J. W. Shimon, G. Evmenenko, P. Dutta, M. A. Iron and M. E. van der Boom, J. Am. Chem. Soc., 2010, 132, 14554 CrossRef CAS.
- M. Altman, M. Rachamim, T. Ichiki, M. Iron, G. Evmenenko, P. Dutta and M. E. van der Boom, Chem.–Eur. J., 2010, 16, 6744 CrossRef CAS; M. Altman, O. V. Zenkina, T. Ichiki, M. A. Iron, G. Evmenenko, P. Dutta and M. E. van der Boom, Chem. Mater., 2009, 21, 4676 CrossRef; M. Altman, O. Zenkina, G. Evmenenko, P. Dutta and M. E. van der Boom, J. Am. Chem. Soc., 2008, 130, 5040 CrossRef; M. Altman, A. D. Shukla, T. Zubkov, G. Evmenenko, P. Dutta and M. E. van der Boom, J. Am. Chem. Soc., 2006, 128, 7374 CrossRef.
- P. K. B. Palomaki, A. Krawicz and P. H. Dinolfo, Langmuir, 2011, 27, 4613 CrossRef CAS.
- L. Motiei, M. Sassi, R. Kaminker, G. Evmenenko, P. Dutta, M. A. Iron and M. E. van der Boom, Langmuir, 2011, 27, 1319 CrossRef CAS.
- J. Choudhury, R. Kaminker, L. Motiei, G. de Ruiter, M. Morozov, F. Lupo, A. Gulino and M. E. van der Boom, J. Am. Chem. Soc., 2010, 132, 9295 CrossRef CAS.
- L. Motiei, Y. Yao, J. Choudhury, H. Yan, T. Marks, M. E. van der Boom and A. Facchetti, J. Am. Chem. Soc., 2010, 132, 12528 CrossRef CAS.
- L. Motiei, R. Kaminker, M. Sassi and M. E. van der Boom, J. Am. Chem. Soc., 2011, 133, 14264 CrossRef CAS; L. Motiei, M. Lahav, A. Gulino, M. A. Iron and M. E. van der Boom, J. Phys. Chem. B, 2010, 114, 14283 CrossRef; L. Motiei, M. Lahav, D. Freeman and M. E. van der Boom, J. Am. Chem. Soc., 2009, 131, 3468 CrossRef.
- L. Motiei, M. Altman, T. Gupta, F. Lupo, A. Gulino, G. Evmenenko, P. Dutta and M. E. van der Boom, J. Am. Chem. Soc., 2008, 130, 8913 CrossRef CAS.
- J. Sagiv and R. Maoz, Adv. Mater., 1998, 10, 580 CrossRef CAS; R. Maoz, S. Matlis, E. DiMasi, B. M. Ocko and J. Sagiv, Nature, 1996, 384, 150 CrossRef.
- Multilayer Thin Films, ed. G. Decher and J. B. Schlenoff, Wiley-VCH, Weinheim, Germany, 2003 CrossRef CAS; N. S. Zacharia, M. Modestino and P. T. Hammond, Macromolecules, 2007, 40, 9523 CrossRef CAS.
- M. Salomaki, I. A. Vinokurov and J. Kankare, Langmuir, 2005, 21, 11232 CrossRef.
- M. Salomaki, T. Laiho and J. Kankare, Macromolecules, 2004, 37, 9585 CrossRef.
- C. Picart, J. Mutterer, L. Richert, Y. Luo, G. D. Prestwich, P. Schaaf, J.-C. Voegel and P. Lavalle, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12531 CrossRef CAS.
- G. de Ruiter, L. Motiei, J. Choudhury, N. Oded and M. E. van der Boom, Angew. Chem., 2010, 122, 4890 CrossRef.
- D. Q. Li, M. A. Ratner, T. J. Marks, C. Zhang, J. Yang and G. K. Wong, J. Am. Chem. Soc., 1990, 112, 7389 CrossRef CAS; R. Yerushalmi, A. Scherz and M. E. van der Boom, J. Am. Chem. Soc., 2004, 126, 2700 CrossRef; S. L. Brandow, T. L. Schull, B. D. Martin, D. C. Guerin and W. J. Dressick, Chem.–Eur. J., 2002, 8, 5363 CrossRef.
- R. J. H. Clark, F. P. Fanizzi, G. Natile, C. Pacifico, C. G. van Rooyen and D. A. Tocher, Inorg. Chim. Acta, 1995, 235, 205 CrossRef CAS.
- S. A. Jasper, R. B. Jones, J. Mattern, J. C. Huffman and L. J. Todd, Inorg. Chem., 1994, 33, 5620 CrossRef CAS.
- P. Espinet, J. M. Martínez-Ilarduya, C. Pérez-Briso, A. L. Casado and M. A. Alonso, J. Organomet. Chem., 1998, 551, 9 CrossRef CAS.
- W. Partenheimer and E. F. Hoy, Inorg. Chem., 1973, 12, 2805 CrossRef CAS; W. Partenheimer, Inorg. Chem., 1972, 11, 743 CrossRef.
- T. Gupta, M. Altman, A. D. Shukla, D. Freeman, G. Leitus and M. E. van der Boom, Chem. Mater., 2006, 18, 1379 CrossRef CAS.
- G. Evmenenko, M. E. van der Boom, S. W. Dugan, J. Kmetko, T. J. Marks and P. Dutta, J. Chem. Phys., 2001, 115, 6722 CrossRef CAS.
- For a discussion regarding the exponential build-up of polyelectrolyte films, see: D. T. Haynie, E. Cho and P. Waduge, Langmuir, 2011, 27, 5700 CrossRef CAS.
- D. T. Haynie, E. Cho and P. Waduge, Langmuir, 2011, 27, 5700 CrossRef CAS.
- C.-P. Li and M. Du, Chem. Commun., 2011, 47, 5958 RSC.
- J. Sarju, J. Arbour, J. Sayer, B. Rohrmoser, W. Scherer and G. Wagner, Dalton Trans., 2008, 5302 RSC.
- G. K. Anderson and M. Lin, Inorg. Synth., 1990, 28, 60 CrossRef CAS.
- D. Drew and J. R. Doyle, Inorg. Synth., 1990, 28, 346 CrossRef CAS.
- P. K. Byers, A. J. Canty, H. Jin, D. Kruis, B. A. Markies, J. Boersma and G. van Koten, Inorg. Synth., 1998, 32, 162 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: UV/Vis absorption spectra (Fig. S1) and AFM images (Fig. S2) of the 6 SPMAs. Synchrotron X-ray reflectivity data of SPMA|Pd(COD)Cl2 (Fig. S3), linear fitting data and molecular densities (Tables S1 and S2). See DOI: 10.1039/c1sc00318f |
| This journal is © The Royal Society of Chemistry 2012 |