DOI:
10.1039/C2RA22483F
(Paper)
RSC Adv., 2012,
2, 12424-12430
The reinforced hydrogel for drug loading: immobilization of single-walled carbon nanotubes in cross-linked polymers via multiple interactions†
Received
11th October 2012
, Accepted 12th October 2012
First published on 12th October 2012
Abstract
We report a facile method to prepare novel carbon nanotube (CNT)-based hydrogels in an aqueous environment. To maximize the bonding between nanotubes and polymers, we exploited several non-covalent interactions like ionic, π–π stacking, hydrophobic, H-bonding, etc. Importantly, the nano-hydrogels swell in both water and organic solvents. The materials showed improvements in various physicochemical properties. The new material is fairly biocompatible, as shown by MTT assay studies. It is also a useful material to load a water-soluble drug or micronutrient such as riboflavin (vitamin B2) or amitriptyline hydrochloride. The releasing study of the drug clearly showed pH responsive behaviour.
1 Introduction
Materials on the nanometer scale are of great contemporary interest for their interesting properties and various applications envisaged for them. During the past few years, great attention has been paid to single-walled carbon nanotubes (SWCNTs) particularly due to their extraordinary mechanical, thermal, and electrical properties.1 The most exciting applications are in the biomedical field,2,3 such as its use as imaging agents and as a near-infrared agent for selective cancer cell destruction, etc. Very recently, we reported the formation of acid responsive microcapsules using SWCNTs for drug delivery applications.3a However, one major bottleneck for its diverse applications in materials science and biotechnology is its poor solubility and low dispersibility in water or organic solvents. This limitation can be overcome by covalent/non-covalent functionalization of single-walled carbon nanotubes (f-SWCNTs).4 Along the same line, we concentrated our focus on developing novel methods for generating better stabilized and improved delivery systems like nanogels based on f-SWCNTs, amphiphilic copolymers based on poly(vinyl imidazole) (PVI) and a cyanuric chloride (CYC)-based cross-linking agent.5
The amphiphilic copolymers that consists of both hydrophobic and hydrophilic segments5 are used extensively in research and technology applications, especially as devices for controlled release of pharmacological substances and biomaterials in tissue engineering.6 One related important issue in design for a specific application in nanotechnology is the appropriate balance between hydrophobic and hydrophilic components, the cross-linking density and the swelling ability.7 It is believed that with the hydrophilic and hydrophobic microdomains formed throughout the network, one could achieve a better regulation of hydrogel morphology and physical properties.8 For example, Yoshida and co-workers have prepared ionene polymer based hydrogels that can act as a dispersant for SWCNT in water.9a In this case, ionic interaction is the main binding force between the polymer and the nanotubes. Prato and co-workers have prepared multiple hydrogen bond interactions with functionalized CNTs that show stable CNT/polymer hybrids.9b Rao and co-workers have prepared SWCNT reinforced supramolecular gels that show good electrical, viscoelastic and near-infrared sensitivity properties.10 However, a weak π–π interaction is the only force to bind the materials. For better binding ability, we have designed the polymer/cross-linking agent and functionalized the nanotubes in such a way that multiple interactions inside the nanogel can take place simultaneously. As depicted in Fig. 1, the designer material contains multiple aromatic rings that impart strong non-covalent interactions11 like π–π stacking with nanotubes, a large number of nitrogen atoms (responsible for strong hydrogen bonding) and quaternization.12 These resulted in strong ionic interactions with carboxylate groups of the CNTs. Another significant advantage in this direction over other methodologies10 is the complete exclusion of hydrophobic organic solvents in the whole process to broaden the scope of applications in the area of biotechnology. To understand the future application area of this kind of novel material, we studied the effect of f-SWCNTs in the swelling, thermal and rheological properties of nanogels.13
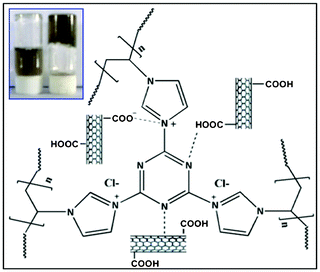 |
| | Fig. 1 Schematic representation of a nanogel. Inset, left: polymer with CNTs, right: nanogel formed by polymer, CNTs and cross-linking agent. | |
2 Experimental
2.1 Materials
Single-walled carbon nanotubes (SWCNTs), vinyl imidazole (VIM) monomer, 2,2′-azo-bis(isobutyronitrile) (AIBN) and D2O (NMR solvent) were purchased from Aldrich, USA, and used as received. All the solvents were purchased from Merck, India.
2.2 Preparation of nanogel
To prepare the nanogel, first the calculated amount of PVI (prepared as per the reported procedure)14a was dissolved in water, further the solution of f-SWCNTs and PVI was mixed together by sonication to obtain the homogeneous f-SWCNTs/PVI solution. The nanocomposites of different weight percentages were prepared by diluting the concentration of polymer in the solution to obtain the final f-SWCNT concentrations of 0.01, 0.015, 0.02, and 0.025 wt% (PS1-4). To this f-SWCNTs/PVI mixture, the 0.05% (w/v) methanol solution of cross-linker cyanuric chloride (0.5 mL) was added, to form the nanogels (Fig. 1).5 The four nanogel samples containing 0.01, 0.015, 0.02 and 0.025% (w/v) were coded as PSG1, PSG2, PSG3, and PSG4, respectively, and studied separately. As a control experiment, polymer gel without f-SWCNTs was also prepared under the same conditions for comparison.
3 Results and discussion
Fig. 1 shows the probable interactions of f-SWCNTs with the polymer in the nanogel. In this case, PVI polymer physically interacts with the f-SWCNTs via π–π stacking formation15 and hydrogen bonding, as confirmed by IR spectroscopy (Fig. 2). In addition, several other non-covalent interactions like hydrogen bonding and ionic interactions are taking place, as concluded from Raman spectra and other data. The inset picture (Fig. 1) clearly shows the visible change after adding the cross-linking agent and nanotubes due to the formation of nanogels.
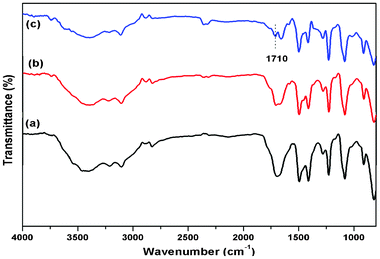 |
| | Fig. 2 FT-IR spectra of (a) polymer gel (without CNTs) (b) nanogel: CNT concentration is 0.01% (PSG1) and (c) nanogel: CNT concentration is 0.025% (PSG4). | |
3.1 FT-IR studies
The FT-IR spectra of PVI and nanocomposites are shown in ESI Fig. S1.† In the case of pure PVI, the peaks at 3100–2950 cm−1 can be assigned to the aromatic and aliphatic C–H stretching vibrations. The absorption peaks at 1650 cm−1 and 1495 cm−1 correspond to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN stretching.16 In the case of f-SWCNTs/PVI without a cross-linker, we see a similar spectrum to the above characteristic peaks, with a small shift in the peak positions. On increasing the concentration of f-SWCNT content in the PVI matrix, we observed a new peak at 1743 cm−1, which was absent in the native polymer,15a indicating the presence of nanotubes in the composite. A substantial broadening from 3100 to 3500 cm−1 (ESI Fig. S2†) supports the hypothesis that the PVI polymer physically interacts through hydrogen bonding.4b The IR spectra of the polymer gel (without CNTs) and nanogels (PSG1 and PSG4) are shown in Fig. 2. A large shift of the peak from 1743 to 1710 cm−1 (for CO stretching frequency) in the nanogel can be attributed to the strong ionic interaction between the carboxylate group of the nanotubes and the quaternary ammonium group of the imidazolium moiety.15b
C and CN stretching.16 In the case of f-SWCNTs/PVI without a cross-linker, we see a similar spectrum to the above characteristic peaks, with a small shift in the peak positions. On increasing the concentration of f-SWCNT content in the PVI matrix, we observed a new peak at 1743 cm−1, which was absent in the native polymer,15a indicating the presence of nanotubes in the composite. A substantial broadening from 3100 to 3500 cm−1 (ESI Fig. S2†) supports the hypothesis that the PVI polymer physically interacts through hydrogen bonding.4b The IR spectra of the polymer gel (without CNTs) and nanogels (PSG1 and PSG4) are shown in Fig. 2. A large shift of the peak from 1743 to 1710 cm−1 (for CO stretching frequency) in the nanogel can be attributed to the strong ionic interaction between the carboxylate group of the nanotubes and the quaternary ammonium group of the imidazolium moiety.15b
3.2 Raman spectroscopic studies
Raman spectra for unmodified pristine SWCNTs, f-SWCNTs, f-SWCNTs/PVI and nanogel (PSG1) are shown in ESI Fig. S3.† The native SWCNTs shows three characteristic bands: radial breathing mode (RBM), G-band and D-band at 174.7, 1597 and 1340 cm−1, respectively.17 The f-SWCNTs show significant changes compared to pristine SWCNTs, the G-band at 1587 cm−1 (lower shift by 10 cm−1), RBM shows two weak peaks at 117 and 170 cm−1, and a predominant D-band at 1357 cm−1.18 This significant change confirms the carboxylic group functionalization on the CNTs, which helps in the preparation of the nanogel.19 The f-SWCNTs/PVI shows the characteristic RBM, G- and D-bands at 171, 1584 and 1344 cm−1, respectively. The nanogel (inset, PSG1) shows a significant change in the G- and D-bands (at 1588 and 1351 cm−1). From the magnified image of the PSG1 Raman spectrum (ESI Fig. S4†), the RBM region shows four weak peaks at 134, 146, 178 and 195 cm−1, respectively.20 These weak peaks with splitting correspond to the individual f-SWCNTs that demonstrate a good dispersion of f-SWCNTs in the nanogel (Table 1).21 The ID/IG ratios of the nanogels (PSG1) showed a higher value than the others due to the defects on the CNT surface.2b This indicates that the defect density in the CNT samples increases by acid and polymer functionalizations. Eklund and co-workers have established that the peak position of RBM can be correlated to the tube diameter, according to a relation: ν (cm−1) = 223.75/d (nm) + Δν (cm−1), which helps to calculate the diameter of the CNTs.22 From the calculation, it was observed that a broader range of diameters resulted for SWCNTs in a nanogel compared to the raw SWCNTs, due to the uneven nature of different kind of interactions (Table 1). The shift in the G- and D-bands with splitting in the RBM region confirms the formation of π–π stacking and hydrogen bonding between the polymer and f-SWCNTs in the gel.
Table 1 Raman spectral data of SWCNTs, f-SWCNTs, f-SWCNTs/PVI and nanogel for comparison
| Type of material |
G-band (cm−1) |
D-band (cm−1) |
I
D/IG ratio |
Diameter (nm) |
| SWCNTs |
1592 |
1340 |
0.476 |
1.36 |
| f-SWCNTs |
1587 |
1357 |
0.692 |
1.38 |
| f-SWCNTs/PVI |
1585 |
1344 |
0.547 |
1.39 |
| PSG1 |
1588 |
1351 |
0.974 |
1.15–1.50 |
The 1H-NMR of PVI shows (ESI Fig. S5†) the characteristic proton signals from the imidazole ring (multiplet at 6.4–7.0 ppm), backbone CH2 (doublet at 1.8–2.2 ppm) group and splitting chain CH group (isotactic, heterotactic and syndiotactic triads).23 The f-SWCNTs/PVI shows a slight downfield shift in the presence of SWCNTs due to the physical interaction of PVI on the surface of f-SWCNTs.14b In the 1H-NMR of nanogels, the signals of the imidazole ring exhibit large downfield shifts due to a positive charge on the imidazole ring formed by the substitution of a CYC cross-linker. In this case, the imidazole ring shows two different types of signals in the aromatic region: at 7.0–7.5 ppm due to the native imidazolium ring proton and the other at 8.4–8.7 ppm for the quaternized imidazolium ring protons.14a
3.4 UV-vis absorption study
The absorption spectrum exhibited the characteristic peaks for PVI polymer at 258 nm, which is in good agreement with reported values (ESI Fig. S6†),14a whereas the CYC cross-linked polymer gel (without CNTs) exhibited two characteristic peaks at 254 nm and 300 nm. The peak at 254 nm with improved intensity is attributed to the presence of the imidazole ring of PVI and a new broad shoulder peak at 300 nm was assigned to the extended conjugation formed by substitution of CYC with the PVI polymer. This substitution makes the π electrons of the imidazole ring of PVI more delocalized compared to PVI, leading to the shifting of the λmax value.24
3.5 Emission study
As seen from the fluorescence spectra (Fig. 3), the addition of f-SWCNTs to the solution of polymer gel resulted in a decrease of the corresponding emission intensity. Here, the PVI based polymer gel showed emission properties (Fig. 3a), with a maximum fluorescence intensity at 425 nm upon λexc at 300 nm. This supports the claim that the polymer gels are self-assembled into aggregates, resulting in the generation of a fluorescence signal of the polymer in an aqueous medium (0.03% w/v).25 On substantial increase of the f-SWCNT concentration sequentially (0.02% w/v) in the polymer gel solution, fluorescence quenching was observed. This quenching was due to the inclusion of f-SWCNTs in self-assembled gelation through π–π interaction.26 Those data clearly indicated that the CNT surface interacted well with the polymer matrix, even at low concentrations.10 The emission intensity at 425 nm showed a good liner relationship of fluorescence intensity with the concentration of nanotubes (coefficient: 0.9724) (Fig. 3b) and one could find that the detection limit was down to 300 μl/3ml (v/v) of f-SWCNTs (0.02% w/v).
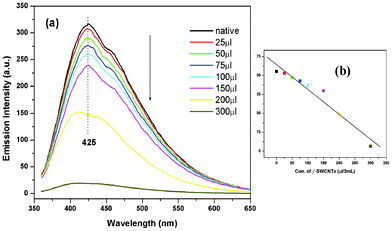 |
| | Fig. 3 (a) Fluorescence spectra of the native gelator (without CNTs) in aqueous solution show a drastic decrease in fluorescence intensity after the incorporation of f-SWCNTs and (b) show the linear fit graph of fluorescence quenching by f-SWCNTs (0.02% w/v) with R2 = 0.9724. | |
3.6 Microscopic studies
The scanning electron microscope (SEM) picture of a dried polymer gel (without CNTs) (Fig. 4a) revealed that the polymers contain small pores, which may be due to the confinement of water molecules.27 However, with the incorporation of f-SWCNTs in the nanogel matrix, agglomeration takes place (Fig. 4b). This suggests that the polymer interacts strongly with f-SWCNTs.10 The individual f-SWCNTs in the nanogel were observed from the high resolution transmission electron microscope image (HR-TEM) (Fig. 4c and ESI Fig. S7†). The incorporated CNTs were observed with an average tube diameter of 25 nm.
The optical microscope images of f-SWCNTs/PVI and the nanogel were shown in Fig. 4(d) and in ESI Fig. S8.† There is somewhat less agglomeration of CNTs at lower concentrations (ESI Fig. S8a†). However, at higher concentration of nanotubes in the polymer matrix some black spots were observed, which are attributed to substantial nanotube agglomerations (ESI Fig. S8b†).28 Interestingly, in the case of the nanogel, multiple layers of agglomerated nanotubes were clearly observed (Fig. 4d).
3.7 Swelling study
The swelling study with deionized water (pH = 7) at 25 °C is plotted in ESI Fig. S9†. All the hydrogels swelled rapidly, reaching equilibrium within 5 min. Due to the presence of the nitrogen atom and charged ions in the back bone of the polymer chain, the materials are hydrophilic in nature.10 The maximum swelling ratio was achieved by the PSG2 nanogel, and was 300% higher compared to the polymer gel, owing to the influence of f-SWCNTs. The swelling behaviours of the polymer gel and PSG2 were also observed in certain selective organic solvents (see ESI Table S1†).29 This observation clearly demonstrates that the incorporation of f-SWCNTs in the polymer improved the swelling ratio of the nanogel compared to the polymer gel (without CNTs).
3.8 Thermal study
The thermal stability of the polymer gel and nanogels was analyzed by the reported inverted tube method.30 The mass of the hydrogel changed into the sol form and finally fell down from the inverted test tube.31 The effect of f-SWCNTs on the thermal stability of the nanogels is shown in ESI Fig. S10.† Importantly, the sol temperature increased from 65 °C in the polymer gel (without CNTs) to 105 °C in the nanogel. This improvement clearly indicates a strong interaction between the nanotubes and the polymer in the presence of a cross-linking agent.
3.9 Thermogravimetric studies
The thermal stability of the f-SWCNTs, homopolymer (PVI) and the nanogel was measured using the thermogravimetric analyser (TGA). The TGA thermograms are shown in Fig. 5. The f-SWCNT thermogram clearly shows the carboxylate functionalization of SWCNTs. The PVI thermogram shows the three-stage degradation temperature. The initial stage was observed at 91 °C with a loss of 10% attributed to the loss of the water in the polymer. The second step of degradation at 410 °C, with a weight loss of 17%, corresponds to the degradation of the organic component. The final stage degradation occurs at 498 °C with a weight loss of 93%, which is due to the decomposition of the main chain. The thermograms of the nanogel with different wt% of CNTs are presented in ESI Fig. S12.† The initial-stage degradation temperature of PSG2 was at 133 °C with a loss of 8% (Fig. 5). This initial decomposition temperature increased up to 42 °C after the incorporation of CNTs into the polymer matrix.32 This increase is because of the hydrogen bonding and charged complex formations between the carboxylate groups on the functionalized SWCNTs, the cross-linking agent and the polymer matrix.31
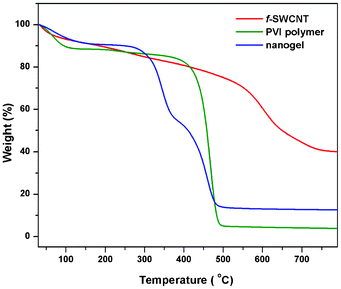 |
| | Fig. 5 TGA of acid functionalized SWCNTs (f-SWCNTs), native polymer (PVI) and nanogel (PSG2). | |
From DSC thermograms (ESI Fig. S13†) the melting temperature (Tm) of the PVI polymer was observed at 115 °C33 and for the polymer gel at 121 °C. In the nanogels, this value shifted to a lower Tm at 114 °C (PSG2) because of the more compact structure caused by the introduction of nanotubes into the imidazole ring.31 The ESI Table S2† summarizes the melting temperatures (Tm), enthalpy changes (ΔH) and calculated activation energy (Ea) from the DSC thermogram of the polymer gel (without CNTs) and the nanogels.10 As seen from the data in the ESI Table S2†, the ΔH and Ea values of the nanogels decreased compared to the polymer gel, due to the strong interactions between the CNTs and the polymer matrix.22
3.10 Rheological study
The storage modulus (G′) and loss modulus (G′′) of the polymer gel and the nanogels were measured using a cone-plate rheometer, as shown in Fig. 6a. The hydrogels with polymer (without CNTs) and nanogel (with CNTs) showed high viscoelastic properties, due to the presence of nitrogen-based heterocyclic structures and charged ions on the polymer chains.9 We were able to observe that nanogels showed higher G′, indicating enhanced stability. When the strain was varied from 0.1 to 100% with 0.5 Hz at 25 °C, the PSG2 showed ∼4 times higher G′ than the polymer gel, indicating enhanced stability and elastic response.10Fig. 6b shows the viscoelastic flow behaviour of the nanogels.
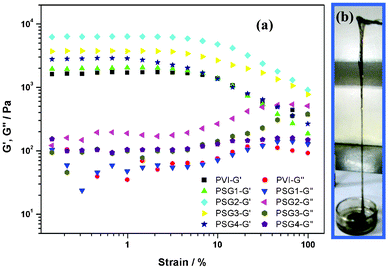 |
| | Fig. 6 Rheograph of (a) polymer gel (without CNTs) and nanogels (b) photograph shows flow behaviour of a nanogel. | |
3.11 MTT assay for biocompatibility
The biocompatibility of the polymer gel and the nanogels was tested using the MTT assay method. Here, the standard procedure was followed for the MTT assay using the 3T3-L1 cells (Fig. 7).27,34 The nanogels and the polymer gel (without CNTs) exhibit good biocompatibility after incubation for 24 h, as presented in Fig. 7. The polymer gel and PSG1 nanogel (0.01% CNTs) show 94% cell viability compare to the control for 24 h, whereas the nanogels of PSG2 (0.015% CNTs) and PSG3 (0.02% CNTs) show above 90% viability.35 However, the PSG4 sample shows only >85%, due to the higher loading, as the aggregated CNTs on the nanogel affects the cell viability.36 Similarly, the MTT assay over 48 h also exhibits above 85% cell viability for the PSG1 and PSG2 nanogels (ESI Fig. S14†). The PSG3 and PSG4 show more than 80% cell viability.37 Recently Lu and co-workers have stated that SWCNTs can be used as a drug delivery vehicle for targeting tumors,38 and the surface functionalized SWCNTs with the highest density of polymer were found to have low toxicity.39 Similarly, Elimelech and co-workers stated that direct contact between the cell and the SWCNT surface is necessary for the inactivation of the cell.40 This finding confirms that the SWCNT surface was well bound in the polymer matrix through multiple interactions. The cell viability study confirms that the nanogels are biocompatible in nature. The biotoxicity of SWCNTs was reduced in the nanogel system and can be used for drug loading and releasing applications in a safe manner.38
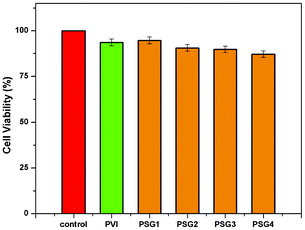 |
| | Fig. 7 MTT assay of PVI-polymer gel (without CNTs) and nanogels (PSG1-4) for biocompatibility over 24 h. | |
3.12 Drug release studies
After confirming that the material is fairly biocompatible and non-toxic in nature, we concentrated on biotechnology applications such as drug loading and releasing studies (Fig. 8 and 9). There are several methods for drug loading either by a covalent or a non-covalent approach.41 We preferred non-covalent methods of drug loading because of the ease of the release process.42 We have chosen riboflavin (vitamin B2) as a model compound because it is water-soluble and fluorescence active. The 0.05% of the riboflavin (vitamin B2) hydrophilic compound was loaded onto the PSG1 sample (0.01% of f-SWCNT) in both neutral pH (pH 7) and acid buffer media (pH 4), simply by the mixing method (ESI†).43 The release kinetics were monitored by using UV-vis absorbance at 264 nm in different time intervals. In neutral pH, the nanogel showed controlled and gradual release of the drug up to 24 h (Fig. 8a).3a In the case of the polymer gel (without CNTs) at neutral pH, the saturation point was achieved in a few hours as presented in ESI Fig S15†. In the case of the nanogel, it showed the saturation point of vitamin B2 release at around 24 h. This controlled and sustainable drug delivery property of the nanogel44 may be due to the formation of multiple interactions of f-SWCNTs with the polymer matrix. Interestingly, in the acid buffer medium, the drug release was faster and reached the saturation point within 3 h (Fig. 8b).41 To understand the clear drug release profile, we have also performed a real drug experiment with an antidepressant drug amitriptyline hydrochloride (AH). The 0.05% of the AH real drug was loaded onto PSG1 nanogel (0.01% of f-SWCNT), and the release kinetics were monitored at 238 nm. In the neutral pH medium, the drug release saturation point was observed at around 24 h (Fig. 9a). However, in the acid buffer medium (pH 4), the drug release was faster (Fig. 9b), as we observed earlier in the vitamin B2 release study. We believe that at acidic pH the ionic interaction of f-SWCNTs in the nanogel was affected; as a result the drug was released faster compared to the case in neutral pH. This finding shows that the nanogels can be used as a drug delivery system for target specific drug release at a designated pH.45 Moreover, this nanogel does not affect the physical properties of the drug even in the gel matrix (Fig. 8a: picture of the nanogel glowing under a UV lamp). These findings may widen the applications from drug delivery to materials science.
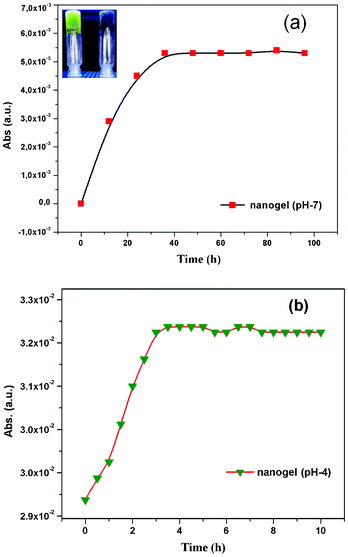 |
| | Fig. 8 UV-vis absorbance at 264 nm of the released riboflavin (vitamin B2) from the nanogel sample (PSG1) at different time intervals (a) in the presence of neutral pH 7; inset: picture of the nanogel glowing by UV lamp, left: with loaded drug, right: without drug and (b) in the presence of acid: pH 4 medium. | |
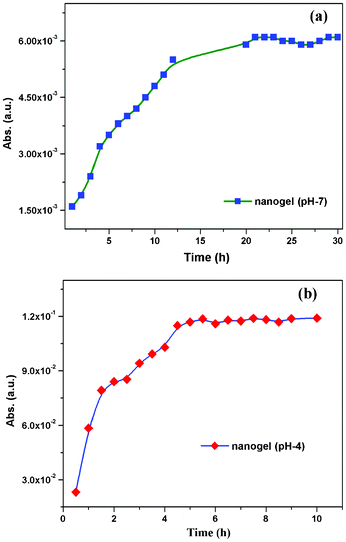 |
| | Fig. 9 UV-vis absorbance at 238 nm of the released amitriptyline hydrochloride (AH) from the nanogel sample (PSG1) at different time intervals (a) in the presence of neutral pH 7 and (b) in the presence of acid: pH 4 medium. | |
4 Conclusions
In conclusion, we have successfully prepared water-based nitrogen rich amphiphilic nanogels. The advantages are two fold. Firstly, the process does not require hydrophobic organic solvents and the gel is water-based. Secondly, the multiple non covalent interactions like H-bonding, ionic, and π–π interactions are taking place simultaneously to maximize the bonding between polymers and nanotubes. The strong bonding resulted in significant improvements in various physicochemical properties in the nanogel, such as rheological properties, sol temperature, swelling behaviour, etc. Clearly the process of hydrogel formation was accelerated in the presence of f-SWCNTs. The Raman study confirms the functionalization of SWCNTs by PVI polymer. The TEM experiment of the nanogel clearly showed the diameter of the nanotubes as approximately 25 nm. The microscopic pictures show the dispersion behaviour of nanotubes in the polymer matrix. The inverted tube experiment showed that the nanogels are more stable than the polymer gel. Better dispersion of the nanotubes leads to a larger storage modulus (rheology study) at low frequencies with improved elasticity.10c Moreover, the TGA and DSC studies of nanogels also showed high initial thermal decomposition and lower melting temperatures compared to the polymer gels. MTT assays showed that the material is fairly biocompatible for biotechnological applications. To understand its potential in drug delivery applications, we loaded a model drug, viz. amitriptyline, and a micronutrient riboflavin (vitamin B2) and studied its releasing behaviour in a neutral as well as an acidic medium. The releasing study of the drug clearly showed pH responsive behaviour, indicating its potential use in targeted drug delivery. We are currently focusing our research on that specific application area, comparing our recently reported pH responsive microcapsules3a and this nanogel system.
Acknowledgements
One of the authors (RMS) is thankful to the Council of Scientific and Industrial Research (CSIR), India (Grant No. 31/6(328)/2010-EMR-I) for fellowship. We sincerely thank Dr Abhijit P. Deshpande, Chemical Engineering, IIT Madras, for helping with the rheological studies. We sincerely thank Dr C. Rose, Biotechnology Lab, CLRI for assistance with the MTT assay.
References
-
(a) S. Iijima and T. Ichihashi, Nature, 1993, 363, 603–605 CrossRef CAS;
(b) Y. Fang, F. Jiang, H. Liu, X. Wub and Y. Lu, RSC Adv., 2012, 2, 6562–6569 RSC;
(c) Q. Zhang, J. Rong and B. Wei, RSC Adv., 2011, 1, 989–994 RSC.
-
(a) Y. Lin, S. Taylor, H. Li, K. A. S. Fernando, L. Qu, W. Wang, L. Gu, B. Zhou and Y.-P. Sun, J. Mater. Chem., 2004, 14, 527–541 RSC;
(b) N. M. Bandaru and N. H. Voelcker, J. Mater. Chem., 2012, 22, 8748–8758 RSC.
-
(a) D. Samanta, R. M. Sankar, S. N. Jaisankar, Md. S. Alam and A. B. Mandal, Chem. Commun., 2011, 47, 11975–11977 RSC;
(b) N. W. S. Kam, M. O. Connell, J. A. Wisdom and H. J. Dai, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11600–11605 CrossRef CAS;
(c) P. Cherukuri, S. M. Bachilo, S. H. Litovsky and R. B. Weisman, J. Am. Chem. Soc., 2004, 126, 15638–15639 CrossRef CAS.
-
(a) J. Liu, A. G. Rinzler, H. Dai, J. H. Hafner, R. K. Bradley, P. J. Boul, A. Lu, T. Iverson, K. Shelimov, C. B. Huffman, F. Rodriguez- Macias, Y. S. Shon, T. R. Lee, D. T. Colbert and R. E. Smalley, Science, 1998, 280, 1253–1256 CrossRef CAS;
(b) S. Dutta, T. Kar, S. Brahmachari and P. K. Das, J. Mater. Chem., 2012, 22, 6623–6631 RSC.
-
(a) C. Lin and I. Gitsov, Macromolecules, 2010, 43, 10017–10030 CrossRef CAS;
(b) B. Fitzpatrick, B. Creran, G. Cooke and V. M. Rotello, Macromol. Chem. Phys., 2012, 213, 1758–1767 CrossRef CAS;
(c) I. W. Hamley, Soft Matter, 2011, 7, 4122–4138 RSC.
- H. Zhang, J. Bei and S. Wang, Biomaterials, 2009, 30, 100–107 CrossRef CAS.
-
(a) D. Missirlis, N. Tirelli and J. A. Hubbell, Langmuir, 2005, 21, 2605–2613 CrossRef CAS;
(b) M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS.
-
(a) Z. Li and T. J. Deming, Soft Matter, 2010, 6, 2546–2551 RSC;
(b) A. Callegari, S. Cosnier, M. Marcaccio, D. Paolucci, F. Paolucci, V. eorgakilas, N. Tagmatarchis, E. Vázquez and M. Prato, J. Mater. Chem., 2004, 14, 807–810 RSC.
-
(a) Y. Misawa, N. Koumura, H. Matsumoto, N. Tamaoki and M. Yoshida, Macromolecules, 2008, 41, 8841–8846 CrossRef CAS;
(b) M. Quintana, H. Traboulsi, A. Llanes-Pallas, R. Marega, D. Bonifazi and M. Prato, ACS Nano, 2012, 6, 23–31 CrossRef CAS.
-
(a) S. K. Samanta, A. Pal, S. Bhattacharya and C. N. R. Rao, J. Mater. Chem., 2010, 20, 6881–6890 RSC;
(b) A. Pal, B. S. Chhikara, A. Govindaraj, S. Bhattacharya and C. N. R. Rao, J. Mater. Chem., 2008, 18, 2593–2600 RSC;
(c) S. Roy and A. Banerjee, RSC Adv., 2012, 2, 2105–2111 RSC.
-
(a) T. Kar, H. F Bettinger, S. Scheiner and A. K. Roy, J. Phys. Chem. C, 2008, 112, 20070–20075 CrossRef CAS;
(b) T. S. Jo, H. Han, L. Mab and P. K. Bhowmik, Polym. Chem., 2011, 2, 1953–1955 RSC;
(c) B. Rybtchinski, ACS Nano, 2011, 5, 6791–6818 CrossRef CAS;
(d) S. Sarkar, S. Niyogi, E. Bekyarova and R. C. Haddon, Chem. Sci., 2011, 2, 1326–1333 RSC.
-
(a) E. B. Anderson and T. E. Long, Polymer, 2010, 51, 2447–2454 CrossRef CAS;
(b) C. Luca, S. Mihailescu and M. Popa, Eur. Polym. J., 2002, 38, 1501–1507 CrossRef CAS;
(c)
D. Samanta, K. Kratz and T. Emrick inGreen Polymerization Methods: Renewable Starting Materials, Catalysis and Waste Reduction, ed. R. T. Mathers and M. A. R. Meier, Wiley-VCH, Weinheim, Germany, 2011, 129-142 Search PubMed.
-
(a) M. Terrones, R. Kamalakaran, T. Seeger and M. Ruhle, Chem. Commun., 2000,(23), 2335–2336 RSC;
(b) S. V. Dommele, K. P. D. Jong and J. H. Bitter, Chem. Commun., 2006,(46), 4859–4861 RSC;
(c) C. Tang, D. Goldberg, Y. Bando, F. Xu and B. Liu, Chem. Commun., 2003,(24), 3050–3051 RSC.
-
(a) A. Srivastava, J. H. Waite, G. D. Stucky and A. Mikhailovsky, Macromolecules, 2009, 42, 2168–2176 CrossRef CAS;
(b) C. Luca, S. Mihailescu and M. Popa, Eur. Polym. J., 2002, 38, 1501–1507 CrossRef CAS;
(c) K-S. Liao, A. Wan, J. D. Batteas and D. E. Bergbreiter, Langmuir, 2008, 24, 4245–4253 CrossRef CAS.
-
(a) B. Zhao, H. Hu and R. C. Haddon, Adv. Funct. Mater., 2004, 14, 71–76 CrossRef CAS;
(b) S. N. Jaisankar, Y. Lakshminarayana and G. Radhakrishnan, Adv. Polym. Technol., 2004, 23, 24–31 CrossRef CAS;
(c) W. Ai, Z. Du, J. Liu, F. Zhao, M. Yi, L. Xie, N. Shi, Y. Ma, Y. Qian, Q. Fan, T. Yu and W. Huang, RSC Adv., 2012 10.1039/c2ra21179c.
- M. Masaki, K. Ogawa and E. Kokufuta, Colloid Polym. Sci., 2009, 287, 1405–1415 CAS.
- M. K. Bayazit, L. S. Clarke, K. S. Coleman and N. Clarke, J. Am. Chem. Soc., 2010, 132, 15814–15819 CrossRef CAS.
- P. Singh, J. Kumar, F. M. Toma, J. Raya, M. Prato, B. Fabre, S. Verma and A. Bianco, J. Am. Chem. Soc., 2009, 131, 13555–13562 CrossRef CAS.
- H. Peng, L. B. Alemany, J. L. Margrave and V. N. Khabashesku, J. Am. Chem. Soc., 2003, 125, 15174–15182 CrossRef CAS.
-
(a) G. R. Dieckmann, A. B. Dalton, P. A. Johnson, J. Razal, J. Chen, G. M. Giordano, E. Munoz, I. H. Musselman, R. H. Baughman and R. K. Draper, J. Am. Chem. Soc., 2003, 125, 1770–1777 CrossRef CAS;
(b) A. Hartschuh and E. J. Sanchez, Phys. Rev. Lett., 2003, 90, 095503–095506 CrossRef.
- Z. Ni, Y. Wang, T. Yu and Z. Shen, Nano Res., 2008, 1, 273–291 CrossRef CAS.
-
(a) A. M. Rao, S. Bandow, E. Richter and P. C. Eklund, Thin Solid Films, 1998, 331, 141–147 CrossRef CAS;
(b) U. D. Venkateswaran, A. M. Rao, E. Richter, M. Menon, A. Rinzler, R. E. Smalley and P. C. Eklund, Phys. Rev. B: Condens. Matter, 1999, 59, 10928–10934 CrossRef CAS.
-
(a) N. Pekel, Z. M. O. Rzaev and O. Guven Macromol, Chem. Phys., 2004, 205, 1088–1095 CAS;
(b) J. Schaefer, E. O. Stejskal and R. Buchdahl, Macromolecules, 1977, 10, 384–405 CrossRef CAS.
-
(a) M. B. Steffensen, E. Hollink, F. Kuschel, M. Bauer and E. E. Simanek, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3411–3433 CrossRef CAS;
(b) N. Karak, B. Roy and B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3994–4004 CrossRef CAS.
- S. K. Mandal, T. Kar, D. Das and P. K. Das, Chem. Commun., 2012, 48, 1814–1816 RSC.
- E. S. Cho, S. W. Hong and W. H. Jo, Macromol. Rapid Commun., 2008, 29, 1798–1803 CrossRef CAS.
- K. Seeni Meera, R. Murali Sankar, S. N. Jaisankar and A. B. Mandal, Colloids Surf., B, 2011, 86, 292–297 CrossRef.
- F. Du, R. C. Scogna, W. Zhou, S. Brand, J. E. Fischer and K. I. Winey, Macromolecules, 2004, 37, 9048–9055 CrossRef CAS.
- B. Adhikari, J. Nanda and A. Banerjee, Chem.–Eur. J., 2011, 17, 11488–11496 CrossRef CAS.
- A. Srivastava, S. Ghorai, A. Bhattacharjya and S. Bhattacharya, J. Org. Chem., 2005, 70, 6574–6582 CrossRef CAS.
-
(a) S. Srinivasan, S. S. Babu, V. K. Praveen and A. Ajayaghosh, Angew. Chem., Int. Ed., 2008, 47, 5746–5749 CrossRef CAS.
- H. Li, D. Q. Wang, H. L. Chen, B. L. Liu and L. Z. Gao, Macromol. Biosci., 2003, 3, 720–724 CrossRef CAS.
- N. Pekel, N. Sahiner, O. Guven and Z. M. O. Rzaev, Eur. Polym. J., 2001, 37, 2443–2451 CrossRef CAS.
-
(a) H. Omidian, K. Park, U. Kandalam and J. G. Rocca, J. Bioact. Compat. Polym., 2010, 25, 483–497 CrossRef CAS;
(b) T. Mosmann and J. Immunol, Methods, 1983, 65, 55–63 CAS.
- A. Bianco, K. Kostarelos, C. D. Partidos and M. Prato, Chem. Commun., 2005, 571–577 RSC.
- H-C. Wu, X. Chang, L. Liu, F. Zhao and Y. Zhao, J. Mater. Chem., 2010, 20, 1036–1052 RSC.
- A. D. Crescenzo, D. Velluto, J. A. Hubbell and A. Fontana, Nanoscale, 2011, 3, 925–928 RSC.
- L. Meng, X. Zhang, Q. Lu, Z. Fei and P. J. Dyson, Biomaterials, 2012, 33, 1689–1698 CrossRef CAS.
-
(a) H. Dumortier, S. Lacotte, G. Pastorin, R. Marega, W. Wu and D. Bonifazi, Nano Lett., 2006, 6, 1522–1528 CrossRef CAS;
(b) C. M. Sayes, F. Liang, J. L. Hudson, J. Mendez, W. H. Guo and J. M. Beach, Toxicol. Lett., 2006, 161, 135–142 CrossRef CAS.
- S. Kang, M. Pinault, L. D. Pfefferle and M. Elimelech, Langmuir, 2007, 23, 8670–8673 CrossRef CAS.
-
(a) M-H. Park, S. S. Agasti, B. Creran, C. Kim and V. M. Rotello, Adv. Mater., 2011, 23, 2839–2842 CrossRef CAS;
(b) S. Bantha, A. V. Kabanov and T. K. Bronich, J. Controlled Release, 2006, 144, 163–174 CrossRef.
-
(a) R. T. Chacko, J. Ventura, J. Zhuang and S. Thayumanavan, Adv. Drug Delivery Rev., 2012, 64, 836–851 CrossRef CAS;
(b) F. Rodrìguez-Llansola, J. F. Miravat and B. Escuder, Chem. Commun., 2011, 47, 4706–4708 RSC.
- Z. Liu, X. Sun, N. Nakayama-Ratchford and H. Dai, ACS Nano, 2007, 1, 50–56 CrossRef CAS.
- T. Fujigaya, T. Morimoto and N. Nakashima, Soft Matter, 2011, 7, 2647–2652 RSC.
- S. Prakash, M. Malhotra, W. Shao, C. Tomaro-Duchesneau and S. Abbasi, Adv. Drug Delivery Rev., 2011, 63, 1340–1351 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Experimental and instrument details; FT-IR, Raman, 1H-NMR and UV-vis spectra of f-SWCNTs/PVI and nanogels; the swelling behaviour and thermal data for all the nanogels. See DOI: 10.1039/c2ra22483f |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here. 