Identification of (phosphine)gold(I) hydrates and their equilibria in wet solutions†
Yu
Tang
and
Biao
Yu
*
State Key Laboratory of Bioorganic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: byu@mail.sioc.ac.cn; Fax: (0086)-21-64166128
First published on 23rd October 2012
Abstract
The elusive gold(I) hydrates [Ph3PAu(OH2)]+TfO− and [((Ph3PAu)2OH)2]2+(TfO−)2 resulting from the most common gold(I) pre-catalyst Ph3PAuOTf have been characterized, leading to disclosure of the equilibria between gold oxo species in wet solutions.
Gold(I) complexes have attracted tremendous attention in recent years, owing to their excellent catalytic activity in a broad range of chemical transformations1 and their potential as therapeutic agents2 and functional materials.3 A theoretical framework has been put forward to rationalize the unique properties of these complexes.1c,4 As homogeneous catalysts, bicoordinate linear complexes LAuX (L is most commonly a tertiary phosphine R3P and X represents a weakly coordinating counteranion such as −OTf, −NTf2, BF4− or SbF6−) are used predominantly as the pre-catalysts. In place of the counteranion X−, unsaturated C–C bonds could coordinate to [LAu]+ and are thus activated for nucleophilic addition.1 A number of the resting intermediates containing LAu–C bonds en route to the final products have been identified recently, supporting the idea that [LAu]+ is the initial catalytic species.1g Recent results indicate that the counteranion X− or an insoluble silver salt (remaining from the preparation of the pre-catalyst) could exert a significant effect on certain transformations.5 However, the catalytic species in these reactions are yet to be investigated.
On the other hand, the gold(I) complexes and their catalyzed reactions are known to be moisture insensitive and are applied mostly without exclusion of moisture or even with water as a solvent. It is therefore important to know whether water reacts with the gold(I) complexes, thus affecting their catalytic activity and turnover rate. In fact, the reaction of [R3PAu]+ with water was reported first in 1980 to afford [(Ph3PAu)3O]+ salt in alkaline or acid media.6,7 Treatment of a relevant [(o-Tol)3PAu)3O]+BF4− (o-Tol = ortho-methylphenyl) with (o-Tol)3PAu+BF4− led to [((o-Tol)3PAu)4O]2+(BF4−)2, which is isolobal to the elusive H4O2+ species.8 Replacement of R3P with more electron-donating N-heterocyclic carbene (NHC) as the ligand has enabled the preparation of IPrAuOH (IPr = N,N′-bis(2,6-diisopropylphenyl)-imidazol-2-ylidene).9 Subjection of IPrAuOH to aqueous HBF4 solution resulted in [(IPrAu)2OH]+BF4.10 Based on these characterized gold(I) oxo species, a reaction pathway of [LAu]+ with water could be proposed, as shown in Fig. 1. In this pathway, the key intermediate [LAu(OH2)]+ has yet to be identified.11 In addition, with the most frequently used ligand R3P, the corresponding LAuOH and (LAu)2OH+ species have not been characterized.
![Proposed equilibria of the gold(i) oxo species in the presence of water. Note: [LAu(OH2)]+ and [(LAu)2OH]+ characterized in the present work represent the first examples of the isolobal series of [H3O]+ with R3P ligands; R3PAuOH and (LAu)2O are yet to be identified.](/image/article/2012/RA/c2ra22282e/c2ra22282e-f1.gif) | ||
| Fig. 1 Proposed equilibria of the gold(I) oxo species in the presence of water. Note: [LAu(OH2)]+ and [(LAu)2OH]+ characterized in the present work represent the first examples of the isolobal series of [H3O]+ with R3P ligands; R3PAuOH and (LAu)2O are yet to be identified. | ||
Preparation of Ph3PAuOTf has become routine practice in our laboratory since the development of a powerful gold(I)-catalyzed glycosylation protocol with glycosyl ortho-alkynylbenzoates as donors.12 Accidentally, crystalline needles (1a, Fig. S1, ESI†) were observed when a batch of newly prepared Ph3PAuOTf in analytical grade CH2Cl2 (containing ∼100 ppm water) was concentrated slowly under reduced pressure. Compound 1a was suspected to be the elusive gold(I) hydrate [LAu(OH2)]+OTf− based on NMR analysis; however, numerous attempts to grow a crystal of 1a suitable for single-crystal X-ray crystallography failed. In fact, complex 1a is hygroscopic and decomposes readily at room temperature. Fortunately, when we switched Ph3P with the bulkier (o-Tol)3P as the ligand, the corresponding homolog 1b was to be found stable and could be obtained as single crystals (see ESI†). X-ray diffraction analysis confirmed it to be the gold(I) hydrate [(o-Tol)3PAu(OH2)]+OTf− (Fig. 2). Thus, the coordination of a water molecule to the (phosphine)gold(I) atom is unambiguously confirmed. In crystal 1b, the Au–O bond length and the P–Au–O angle are 2.069(6) Å and 174.7(2)°, respectively.
 | ||
| Fig. 2 ORTEP diagram of complexes 1b, 2a, and 2b with 30% probability ellipsoids (hydrogen atoms and the TfO− in 2a are omitted for clarity). Key bond lengths (Å) and angles (°) for 1b: Au–O 2.070(7), Au–P 2.208(2), P–Au–O 174.6(3); 2a: Au1–O1 2.041(4), Au2–O2 2.033(5), Au1–O1–Au2 126.3(4), Au3–O2–Au4 127.7(6); 2b: Au1–O1 2.047(3), Au2–O1 2.070(3), Au3–O2 2.070(3), Au4–O2 2.042(2), Au1–Au3 3.397, Au1–Au4 3.486, Au2–Au3 3.1035, Au2–Au4 3.2798(3), Au1–O1–Au3 129.09(16), Au2–O2–Au4 123.34(14). | ||
An attempt at recrystallization of complex 1a in CH2Cl2/i-Pr2O led to colorless tetragonal dipyramid shaped crystals (2a) and colorless rectangular shaped crystals (2b) (Fig. S7, ESI†) (Scheme 1). These crystals were separated manually and determined to be the stereoisomers of [((Ph3PAu)2OH)2]2+(TfO−)2 (Fig. 2). We later found that the formation of these products was time-dependent in the presence of 5 Å molecular sieves in the initial reaction system. Thus, stirring the mixture of Ph3PAuCl and AgOTf (with 5 Å MS) for 1 h followed by crystallization led to 2a/2b in yields of up to 65%; while stirring for 6 h followed by addition of i-Pr2O resulted in a white precipitate, which crystallized in various solvents to provide [(Ph3PAu)3O]+TfO− (3) (Fig. S12 and S13, ESI†).13
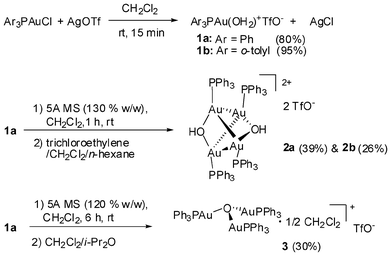 | ||
| Scheme 1 Preparation of the gold(I) oxo complexes 1–3. | ||
Both complex 2a and 2b are tetranuclear dimers with a quasi-tetrahedral array of the four gold atoms in crystals. Complex 2a has a highly symmetrical structure (point group S4). The Au–Au distance in each monomer unit is 3.65 Å, indicating the absence of any significant aurophilic interaction. The Au–Au bonds between the two monomers units are 3.1939(5) Å and 3.2191(6) Å, showing a typical aurophilic interaction. Complex 2b contains two dinuclear monomer units with different geometries. The Au–Au distances in and between the monomers are 3.71 Å/3.67 Å and 3.1035(3) Å/3.2798(3) Å, respectively, showing again that aurophilic interactions occur only between the two monomer units.
Previous structural studies of trigold oxonium cations [(R3P)Au3O]+ reveal a similar tetrahedral contact of the monomers with trimethylphosphine ligands (R = Me), whereas all other salts of this type (R = Ph etc.) form rectangular dimers, or are monomeric. Theoretical studies13c show that for bulkier ligands such as triphenylphosphine (R = Ph), the rectangular structure is clearly favored, due to steric intermonomer repulsion. Our results here show that in the case of gold oxo complexes 2a and 2b, a “tetrahedral” dimer could be formed even with bulkier ligands such as triphenylphosphine. These structures provide new cases of isolobal species with an isostructural core.
It is noted that hydrogen bonds between the hydroxyl group and the CF3SO3− anion were observed in all three complexes. This interaction may facilitate crystal growth. In fact, all attempts to obtain crystals of the (phosphine)gold(I) complexes with non-coordinating anions such as BF4− and SbF6− failed.14 In addition, the present dimeric (phosphine)gold(I) hydrates 2a/2b are different from the NHC based hydroxo-bridged dinuclear cation [((NHC)Au)2OH]+, which exists as a monomer in crystals.10b Another related example is the di[gold(I)]halonium salts X[Au(PR3)]2+ (X = Cl, Br, I). With large anion SbF6− they exist as tetranuclear dications, while with small anions such as BF4− and ClO4−, they occur as a dinuclear monomer.15 These results indicate that the association between polyaurated onium salts depends on both ligand and anion, and hydrogen bonding may also play an important role.
The 31P and 1H NMR spectra of gold(I) oxo complexes 1a, 2a/2b and 3 were measured (see ESI†). Interestingly, the 31P signals shift upfield gradually as the number of [Ph3PAu]+ cations coordinated with oxygen increases (Fig. 3). The 1H NMR signals of 1a, 2a/2b, and 3 in the aromatic region have different shapes and positions (Fig. S16, ESI†), despite the fact that they contain Ph3P as the sole ligand. Both the 31P and 1H NMR spectra of 2a and 2b are identical, implying that they adopt the same solution structure.
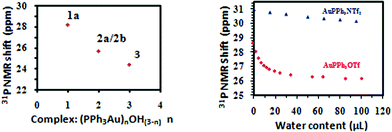 | ||
| Fig. 3 31P NMR chemical shifts of the gold oxo complexes (1a, 2a/2b, and 3) and Ph3PAuOTf/Ph3PAuNTf2 in CD2Cl2 in the presence of varied amounts of water. | ||
Based on the data of the authentic samples, the reaction of water with Ph3PAuOTf and Ph3PAuNTf2,14 two widely used homogeneous gold(I) pre-catalysts,1 was investigated by 31P and 1H NMR spectrometry. Experimentally, water was added gradually to a freshly prepared CD2Cl2 solution of Ph3PAuOTf (or Ph3PAuNTf2), and the 31P and 1H NMR spectra were recorded after each addition (Fig. S18–21, ESI†).
Key results obtained from these experiments include: (a) in all cases, 31P NMR spectra showed only one single sharp signal, indicating the existence of a rapid equilibrium between interchanging species, as is also suggested by the crystallization experiments under various conditions (see ESI†). (b) For Ph3PAuOTf bearing weakly coordinating anion TfO−, [Ph3PAu(OH2)]+TfO− (1a) formed readily when small amounts of water were introduced into the solution. As the amount of water increased, [((Ph3PAu)2OH)2]2+(TfO−)2 (2) formed gradually, accompanied by the generation of H3O+:
| [Ph3PAu]+ + H2O → [Ph3PAu(OH2)]+ |
| 2[Ph3PAu(OH2)]+ → [(Ph3PAu)2OH]+ + H3O+ |
The coordination of water to [Ph3PAu]+ in solution was also observed by 1H NMR measurement (see ESI†). For Ph3PAuOTf in CD2Cl2 containing 1.0 eq water, the water signal shifted downfield from 1.52 (in pure CD2Cl2) to 4.56 ppm (Fig. S17, ESI†). In contrast, the water (1.0 eq) signal in AuPPh3NTf2 solution showed up only slightly downfield at 1.69 ppm. These results strongly suggest that the coordination of [Ph3PAu]+ with NTf2− is much more favorable than with water in solution.
The existence of dynamic equilibria rather than a single species in solution was further supported by crystallization experiments under various conditions (Table S2, ESI†). Complex 1a was formed only in non-coordinating solvents, whereas in oxygen-containing solvents the major product was complex 2. Interestingly, recrystallization of complex 2 in wet non-coordinating solvents led to complex 3, with only a small amount of complex 2 remaining. These results show clearly that the reaction equilibrium is strongly affected by the nature of the solvent.
The role of molecular sieves (which is always used in the glycosylation reaction to remove moisture)12 in the present reaction was briefly investigated. When 5 Å MS was added to a mixture of AuPPh3Cl and AgOTf (1.0 eq) in CH2Cl2, the 31P signals shifted gradually upfield from 28.04 ppm to 25.95 ppm as the stirring time went on (Fig. S22, ESI†). Crystallization experiments confirmed that the major product changed gradually from 1a to 2a and then to 3 (Table S2, ESI†). Apparently, the molecular sieves serve as a mild base, which slowly neutralize the acid generated in situ, leading to a shift in equilibrium.
In conclusion, the formation of (phosphine)gold(I) hydrates could be a ready process in the presence of water. This finding provides a good explanation for the stability of the gold(I) complexes known to chemists, so that LAuNTf2,14 [LAu-triazole]X17 and LAu(NCMe)X,1e,18 which resist hydration, have been found stable, while the readily hydrolysable LAuX (X− = −OTf, BF4− and SbF6−)14 are found unstable. More importantly, the present finding shall afford a new visual angle for looking into the mechanism of gold(I) catalysis in certain reactions. Thus, the dramatic difference between the catalytic behaviour of Ph3PAuOTf and Ph3PAuNTf2 might correlate to their disparate reactivities toward moisture;5a the previous proposal of new gold(I) species based on the 31P NMR signal might have overlooked this hydration process.5a Additionally, awareness of the hydration process of gold(I) complexes will also be helpful in understanding their biological and material properties.
Acknowledgements
We thank the National Natural Science Foundation of China (20932009 and 20921091) and the Ministry of Science and Technology of China (2012ZX09502-002) for funding and X. B. Leng and J. Sun for X-ray crystallographic analysis.References
- For selected reviews, see: (a) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef; (b) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef; (c) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395 CrossRef CAS; (d) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS; (e) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef CAS; (f) S. P. Nolan, Acc. Chem. Res., 2011, 44, 91 CrossRef CAS; (g) L.-P. Liu and G. B. Hammond, Chem. Soc. Rev., 2012, 41, 3129 RSC.
- I. Ott, Coord. Chem. Rev., 2009, 253, 1670 CrossRef CAS.
- J. C. Lima and L. Rodriguez, Chem. Soc. Rev., 2011, 40, 5442 RSC.
- (a) P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412 CrossRef; (b) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC; (c) H. G. Raubenheimer and H. Schmidbaur, Organometallics, 2012, 31, 2507 CrossRef CAS.
- (a) D. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedov, H. Zhang, X. Liu, J. L. Petersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012 Search PubMed; (b) H. Schmidbaur and A. Schier, Z. Naturforsch., B: J. Chem. Sci., 2011, 66b, 329 Search PubMed.
- A. N. Nesmeyanov, E. G. Perevalova, Yu. T. Struchkor, M. Yu. Antipin, K. I. Grandberg and V. P. Dyadchenko, J. Organomet. Chem., 1980, 201, 343 CrossRef CAS.
- This type of gold(I)-oxo complexes have been used in catalysis, see for example: (a) B. D. Sherry and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 15978 CrossRef CAS; (b) B. D. Sherry, L. Maus, B. N. Laforteza and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 8132 CrossRef CAS; (c) R. L. LaLonde, W. E. Brenzovich, D. Benitez, E. Tkatchouk, K. Kelley, W. A. Goddard III and F. D. Toste, Chem. Sci., 2010, 1, 226 RSC.
- (a) H. Schmidbaur, S. Hofreiter and M. Paul, Nature, 1995, 377, 503 CrossRef CAS; (b) K. Nomiya, T. Yoshida, Y. Sakai, A. Nanba and S. Tsuruta, Inorg. Chem., 2010, 49, 8247 CrossRef CAS.
- S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742 RSC.
- (a) S. Gaillard, J. Bosson, R. S. Ramón, P. Nun, A. M. Z. Slawin and S. P. Nolan, Chem.–Eur. J., 2010, 16, 13729 CrossRef CAS; (b) R. S. Ramón, S. Gaillard, A. Poater, L. Cavallo, A. M. Z. Slawin and S. P. Nolan, Chem.–Eur. J., 2011, 17, 1238 CrossRef CAS.
- G. N. Khairallah, R. A. J. O'Hair and M. I. Bruce, Dalton Trans., 2006, 3699 RSC.
- (a) Y. Li, Y. Yang and B. Yu, Tetrahedron Lett., 2008, 49, 3604 CrossRef CAS; (b) Y. Zhu and B. Yu, Angew. Chem., Int. Ed., 2011, 50, 8329 CAS; (c) J. Zhang, H. Shi, Y. Ma and B. Yu, Chem. Commun., 2012, 48, 8679 RSC; (d) B. Yu, J. Sun and X. Yang, Acc. Chem. Res., 2012, 45, 1227 Search PubMed.
- (a) Y. Yang, V. Ramamoorthy and P. R. Sharp, Inorg. Chem., 1993, 32, 1946 CrossRef CAS; (b) K. Angermaier and H. Schmidbaur, Inorg. Chem., 1994, 33, 2069 CrossRef CAS; (c) S.-C. Chung, S. Krüger, H. Schmidbaur and N. Rösch, Inorg. Chem., 1996, 35, 5387 CrossRef CAS.
- N. Mézailles, L. Ricard and F. Gagosz, Org. Lett., 2005, 7, 4133 CrossRef.
- (a) R. Uson, A. Laguna and M. V. Castrillo, Synth. React. Inorg. Met.-Org. Chem., 1979, 9, 317 CAS; (b) A. Hamel, N. W. Mitzel and H. Schmidbaur, J. Am. Chem. Soc., 2001, 123, 5106 CrossRef CAS; (c) H. Schmidbaur, A. Hamel, N. W. Mitzel, A. Schier and S. Nogai, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4916 CrossRef CAS.
- S. Antoniotti, V. Dalla and E. Duñach, Angew. Chem., Int. Ed., 2010, 49, 7860 CrossRef CAS.
- H. Duan, S. Sengupta, J. L. Petersen, N. G. Akhmedov and X. Shi, J. Am. Chem. Soc., 2009, 131, 12100 CrossRef CAS.
- E. Herrero-Gómez, C. Nieto-Oberhuber, S. López, J. Benet-Buchholz and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 5455 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, and NMR spectra for new compounds. Crystallographic data for compounds 1b, 2a, 2b, 3 H2O, and 3 1/2CH2Cl2. CCDC reference numbers 892405–892409. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra22282e |
| This journal is © The Royal Society of Chemistry 2012 |