Construction of tetrahydro-β-carboline skeletons via Brønsted acid activation of imide carbonyl group: syntheses of indole alkaloids (±)-harmicine and (±)-10-desbromoarborescidine-A†
Selvaraj
Mangalaraj
and
Chinnasamy Ramaraj
Ramanathan
*
Department of Chemistry, Pondicherry University, Puducherry – 605 014, India. E-mail: crrnath.che@pondiuni.edu.in; Fax: +91-413-2656740; Tel: +91-413-2654416
First published on 25th September 2012
Abstract
Indole, a π-nucleophile, reacts with Brønsted acid activated imide carbonyl group in an intramolecular fashion via a 6-exo-trig cyclization, to deliver a condensed tetrahydro-β-carboline unit. This methodology is effectively applied to assemble the tetrahydro-β-carboline skeleton containing alkaloids such as harmicine and 10-desbromoarborescidine-A.
Terpene indole alkaloids, with a tetrahydro-β-carboline (THBC) subunit, have been isolated from various plants and animals.1 The structural diversity of these complex natural products account for the plethora of biological activities.2 For example, the alkaloid (+)-yohimbine has been used to treat erectile dysfunction;3 (+)-vincamine, a vasodilator, has been used to increase the blood flow in brain;4 and the alkaloid (−)-reserpine, has been used to treat the hypertension5 (Fig. 1).
 | ||
| Fig. 1 Indole alkaloids. | ||
This synthetically and biologically important structural class demands the development of simple and efficient synthetic methodologies. Usually the assemblage of this heterocyclic system employs reactions such as the Pictet–Spengler reaction,6 Bischler–Napieralski reaction,7N-acyliminium ion cyclization,8 cycloaddition reaction,9 and keteniminium Pictet–Spengler reaction.10 These methods generally require either harsh conditions and/or several steps.
Matching the electrophilicity of the imide carbonyl carbon through Lewis acid or Brønsted acid activation with the nucleophilicity of π-nucleophiles, such as aromatic systems, may result in C–C bond formation. Recently, we have demonstrated this concept to effect the intramolecular cyclization of phenethylimides via imide carbonyl activation to generate tetrahydroisoquinolines.11 This observation prompted us to investigate the feasibility of assembling the tetrahydro-β-carboline skeleton through imide carbonyl activation.
Accordingly, the phthalimido derivative of tryptamine 1a was treated with 2 equivalents of trifluoromethanesulfonic acid (TfOH) in dichloromethane, which failed to generate the cyclized product. Though, the theoretically predicted global nucleophilicity of indole was found to be higher than anisole,12 it failed to react intramolecularly with the activated imide carbonyl group. However, we earlier witnessed, the successful cyclization of methoxy substituted benzene through imide carbonyl activation using acids at room temperature.11b This difference may be due to the existence of protonated species I in the presence of TfOH (pKa = −14), similar to the protonated indole (pKa = −3.6) existing in the strong acid medium (Fig. 2).13
 | ||
| Fig. 2 Protonation of indole moiety. | ||
When the reaction was performed with TfOH (4 equiv.) in dichloromethane only 22% of cyclized product 2 was produced, even after 24 h. Whereas the imide 1a underwent cyclization on treatment with TfOH (4 equiv.) in the presence of powdered and freshly dried 4 Å molecular sieves to afford the expected tetrahydro-β-carboline 2 with increased yield (29%) in a shorter reaction time (12 h). The THBC derivative 2 with 81% yield was realized when the imide 1a was treated with TfOH (10 equiv.) in the presence of 4 Å molecular sieves (Scheme 1). In the absence of molecular sieves, the substrate 1a produced the THBC 2 in 44% yield upon treatment with TfOH (10 equiv.) in dichloromethane. Hence, we presume that the molecular sieves may be facilitating the formation of the fused cyclic acyl iminium ion II via hydroxy lactam formation. Though the starting material was completely consumed, the isolation of the cyclized hydroxy lactam 2 became cumbersome due to its polar nature. Hence, in situ reduction of the hydroxy lactam using NaBH4/CF3COOH14 after neutralizing the reaction mixture with solid NaHCO3 followed by neutral alumina column chromatography, furnished the heterocyclic lactam 2a in 83% yield (Scheme 1).
 | ||
| Scheme 1 Cyclization of the phthalimido derivative of tryptamine. | ||
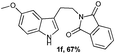
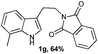
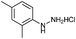
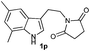
Successful cyclization of the phthalimide derivative of tryptamine prompted us to investigate the electronic effect of substituents on the internal nucleophile (indole ring) in this reaction. The imide derivatives of tryptamines are usually prepared by direct condensation of substituted tryptamines with anhydrides in refluxing toluene.10a However, the cost of substituted tryptamines intrigued us to develop a simple methodology to synthesize the imide derivatives of substituted tryptamines. Thus, we have adopted the methodology that has been utilized to prepare N,N-dimethyltryptamines.15 The imide derivatives of substituted tryptamines were produced in moderate yield from the imide derivative of 4-amino-1-butanol via Swern oxidation, in situ aldehyde protection using propane-1,3-diol, followed by indolization using 4% sulphuric acid (Table 1, entries 1–7). Here, the primary amine, protected as an imide, was intact during this sequence of reactions. This methodology can be used as a general tool to synthesize the imide derivative of tryptamines.
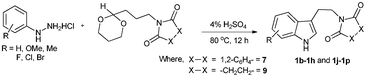
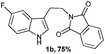
| Entry | Substituted phenylhydrazine hydrochloride | Phthalimide derivative of tryptamines | Succinimide derivative of tryptamines |
|---|---|---|---|
| a Reaction conditions: 7 or 9 (2.0 mmol), substituted phenyl hydrazine hydrochloride (2.0 mmol), 4% H2SO4 (100 ml), 80 °C, 12 h. | |||
| 1 |

|
|
|
| 2 |

|
|

|
| 3 |

|

|

|
| 4 |

|

|

|
| 5 |

|
|

|
| 6 |

|
|

|
| 7 |
|

|

|
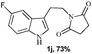
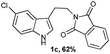
A wide substrate scope for this cyclization strategy can be envisaged as most of the phthalimide derivatives of substituted tryptamines produced the expected tetrahydro-β-carboline derivatives, 2b–h (Table 2, entries 1–7). Under this cyclization conditions, the indole ring, with both electron releasing and withdrawing functional groups, furnished the expected pentacyclic lactams in moderate to good yields (Table 2, entries 1–7).
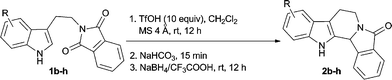














Most of the indole alkaloids that occur in nature possess a tetrahydro-β-carboline skeleton fused with pyrrolidine or piperidine units. Therefore, the alicyclic imide derivatives of tryptamines were subjected to the cyclization reaction using TfOH/MS 4 Å followed by reduction using NaBH4/MeOH in dichloromethane. As expected, the condensed tetracyclic or pentacyclic heterocycles were generated in good yields (Table 3, entries 1–10).



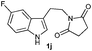
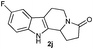



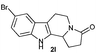


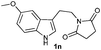








The formation of these tetrahydro-β-carboline derivatives was unambiguously confirmed by IR, NMR (1H and 13C), and HRMS data. The structure of the compounds 2, 2e, 2h, 2o and 2q were further confirmed by single crystal X-ray crystallography16 (Fig. 3). Notably the cis-cyclohexane-1,2-dicarboximide derivative of tryptamine 1q underwent diastereoselective cyclization to produce one diastereomer, 2q (Table 3, entry 9) in which the hydrogens on the fused carbons are cis to each other as evident from the single crystal structural analysis (Fig. 3).
 | ||
| Fig. 3 ORTEP diagrams of compounds 2, 2e, 2h, 2o and 2q. | ||
The synthetic efficiency of this methodology was further exemplified through the syntheses of indole alkaloids such as (±)-harmicine,17 isolated from Kopsia griffithii, and (±)-10-desbromoarborescidine-A,18 isolated from Pseudodistoma arborescens in racemic form from the corresponding imide derivatives of tryptamine. The successful assemblage of (±)-harmicine and (±)-10-desbromoarborescidine-A was effected by the initial condensation of tryptamine with succinic anhydride and glutaric anhydride followed by TfOH treatment and reduction using NaBH4/MeOH. The precursors 2i and 2r were produced in 82% and 87% yields respectively from imides 1i and 1r. The precursors 2i and 2r, upon reduction using LiAlH4, effectively furnished the natural products (±)-harmicine 4 and (±)-10-desbromoarborescidine-A 5 in 78% and 63% yields respectively (Scheme 2).
 | ||
| Scheme 2 Synthesis of (±)-harmicine and (±)-10-desbromoarborescidine-A. | ||
In conclusion, we have demonstrated the Brønsted acid assisted 6-exo-trig cyclization of imide derivatives of tryptamine through imide carbonyl activation to assemble the tetrahydro-β-carboline derivatives. The utility of this methodology was exemplified by the successful and short synthesis of (±)-harmicine and (±)-10-desbromoarborescidine-A. Using this methodology as a key step, the synthesis of indole alkaloids with further structural complexity are progressing in this laboratory.
Acknowledgements
We thank the Council of Scientific and Industrial Research, India, and the Department of Science and Technology, India for financial support. S. M. thanks the University Grants Commission, India for the fellowship. We are grateful to the Central Instrumentation Facility, Pondicherry University for IR and NMR (1H and 13C) data. Single Crystal X-ray facility (DST-FIST sponsored), Department of Chemistry, Pondicherry University is gratefully acknowledged. We thank SAIF, IITM, Chennai for HRMS data.References
- Dictionary of alkaloids, ed. J. Buckingham, K. H. Baggaley, A. D. Roberts and L. F. Szabo, 2nd edn, CRC Press, USA, 2010 Search PubMed.
- (a) N. Sunder-Plassmann, V. Sarli, M. Gartner, M. Utz, J. Seiler, S. Huemmer, T. U. Mayer, T. Surrey and A. Giannis, Bioorg. Med. Chem., 2005, 13, 6094 CrossRef CAS; (b) G. N. Maw, C. M. N. Allerton, E. Gbekor and W. A. Million, Bioorg. Med. Chem. Lett., 2003, 13, 1425 CrossRef CAS; (c) For biochemical pharmacological functions see the review: R. Cao, W. Peng, Z. Wang and A. Xu, Curr. Med. Chem., 2007, 14, 479 CrossRef CAS; (d) G. Bringmann, D. Feineis, R. Bruckner, M. Blank, K. Peters, E.-M. Peters, H. Reichmann, B. Janetzky, C. Grote, H.-W. Clement and W. Wesemann, Bioorg. Med. Chem., 2000, 8, 1467 CrossRef CAS; (e) R. Di Fabio, F. Micheli, G. Alvaro, P. Cavanni, D. Donati, T. Gagliardi, G. Fontana, R. Giovannini, M. Maffeis, A. Mingardi, M. E. Tranquillini and G. Vitulli, Bioorg. Med. Chem. Lett., 2007, 17, 2254 CrossRef CAS; (f) J. I. Trujillo, M. J. Meyers, D. R. Anderson, S. Hegde, M. W. Mahoney, W. F. Vernier, I. P. Buchler, K. K. Wu, S. Yang, S. J. Hartmann and D. B. Reitz, Bioorg. Med. Chem. Lett., 2007, 17, 4657 CrossRef CAS; (g) L. Gupta, K. Srivastava, S. Singh, S. K. Puri and P. M. S. Chauhan, Bioorg. Med. Chem. Lett., 2008, 18, 3306 CrossRef CAS.
- E. Ernst and M. H. Pittler, J. Urol., 1998, 159, 433 CrossRef CAS.
- A. Vas and B. Gulyas, Med. Res. Rev., 2005, 25, 737 CrossRef CAS.
- G. Lemieux, A. Davignon and J. Genest, Can. Med. Assoc. J., 1956, 74, 522 CAS.
- (a) E. D. Cox and J. M. Cook, Chem. Rev., 1995, 95, 1797 CrossRef CAS; (b) M. Rueping and C. M. R. Volla, RSC Adv., 2011, 1, 79 RSC; (c) J. Stockigt, A. P. Antonchick, F. Wu and H. Waldman, Angew. Chem., Int. Ed., 2011, 50, 8538 CrossRef CAS; (d) E. Airiau, N. Girard, A. Mann, J. Salvadori and M. Taddei, Org. Lett., 2009, 11, 5314 CrossRef CAS; (e) E. E. Van Tamelen and L. K. Oliver, J. Am. Chem. Soc., 1970, 92, 2136 CrossRef CAS; (f) S. B. Mandal, V. S. Giri, M. S. Sabeena and S. C. Pakrashi, J. Org. Chem., 1988, 53, 4236 CrossRef CAS; (g) L. E. Overman and T. C. Malone, J. Org. Chem., 1982, 47, 5297 CrossRef CAS; (h) T. Ito, M. Kitajima and H. Takayama, Tetrahedron Lett., 2009, 50, 4506 CrossRef CAS; (i) M. J. Wanner, R. N. A. Boots, B. Eradus, R. de Gelder, J. H. van Maarseveen and H. Hiemstra, Org. Lett., 2009, 11, 2579 CrossRef CAS; (j) H. Liu and A. Dömling, J. Org. Chem., 2009, 74, 6895 CrossRef CAS; (k) J. Ma, W. Yin, H. Zhou, X. Liao and J. M. Cook, J. Org. Chem., 2009, 74, 264 CrossRef CAS; (l) H. J. Kumpaty, M. L. V. Linn, M. S. Kabir, F. H. Forsterling, J. R. Deschamps and J. M. Cook, J. Org. Chem., 2009, 74, 2771 CrossRef CAS; (m) M. Barbero, S. Bazzi, S. Cadamuro and S. Dughera, Tetrahedron Lett., 2010, 51, 6356 CrossRef CAS; (n) W. Wang, E. Herdtweck and A. Dömling, Chem. Commun., 2010, 46, 770 RSC; (o) T. Arai, M. Wasai and N. Yokoyama, J. Org. Chem., 2011, 76, 2909 CrossRef CAS; (p) C. Sanaboina, S. Jana, S. Chidara, B. Patro, G. B. Raolji and L. Eppakayala, Tetrahedron Lett., 2012, 53, 5027 CrossRef CAS.
- (a) L. S. Santos, C. Theoduloz, R. A. Pilli and J. Rodriguez, Eur. J. Med. Chem., 2009, 44, 3810 CrossRef CAS; (b) W. A. da Silva, M. T. Rodrigues Jr, N. Shankaraiah, R. B. Ferreira, C. K. Z. Andrade, R. A. Pilli and L. S. Santos, Org. Lett., 2009, 11, 3238 CrossRef CAS; (c) M.-S. Chern, Y.-K. Shih, P. M. Dewang and W.-R. Li, J. Comb. Chem., 2004, 6, 855 CrossRef CAS; (d) A. Spaggiari, L. C. Blaszczak and F. Prati, Org. Lett., 2004, 6, 3885 CrossRef CAS; (e) T.-L. Ho and Q.-x. Lin, Tetrahedron, 2008, 64, 10401 CrossRef CAS; (f) Atta-ur-Rahman and N. Waheed, Tetrahedron Lett., 1977, 18, 4101 CrossRef; (g) T. Fujii, S. Yoshifuji and H. Ito, Chem. Pharm. Bull., 1988, 36, 3348 CrossRef CAS.
- (a) I. T. Raheem, P. S. Thiara, E. A. Peterson and E. N. Jacobsen, J. Am. Chem. Soc., 2007, 129, 13404 CrossRef CAS; (b) C. A. Holloway, M. E. Muratore, R. I. Storer and D. J. Dixon, Org. Lett., 2010, 12, 4720 CrossRef CAS; (c) Atta-ur-Rahman, M. Ghazala, N. Sultana and M. Bashir, Tetrahedron Lett., 1980, 21, 1773 CrossRef CAS.
- (a) A. Padwa, L. S. Beall, T. M. Heidelbaugh, B. Liu and S. M. Sheehan, J. Org. Chem., 2000, 65, 2684 CrossRef CAS; (b) G. Poissonnet, M. H. Theret-Bettiol and R. H. Dodd, J. Org. Chem., 1996, 61, 2273 CrossRef CAS.
- Y. Zhang, R. P. Hsung, X. Zhang, J. Huang, B. W. Slafer and A. Davis, Org. Lett., 2005, 7, 1047 CrossRef CAS.
- (a) J. Selvakumar, A. Makriyannis and C. R. Ramanathan, Org. Biomol. Chem., 2010, 8, 4056 RSC; (b) J. Selvakumar and C. R. Ramanathan, Org. Biomol. Chem., 2011, 9, 7643 RSC.
- S. Pratihar and S. Roy, J. Org. Chem., 2010, 75, 4957 CrossRef CAS.
- (a) F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS; (b) F. G. Bordwell, G. E. Drucker and H. E. Fried, J. Org. Chem., 1981, 46, 632 CrossRef CAS; (c) R. L. Hinman and J. Lang, J. Am. Chem. Soc., 1964, 86, 3796 CrossRef CAS; (d) A. Kulkarni, W. Zhou and B. Torok, Org. Lett., 2011, 13, 5124 CrossRef CAS.
- I. Osante, M. I. Collado, E. Lete and N. Sotomayor, Eur. J. Org. Chem., 2001, 1267 CrossRef CAS.
- C. Chen, C. H. Senanayake, T. J. Bill, R. D. Larsen, T. R. Verhoeven and P. J. Reider, J. Org. Chem., 1994, 59, 3738 CrossRef CAS.
- For details see the ESI†.
- (a) T. S. Kam and K. M. Sim, Phytochemistry, 1998, 47, 145 CrossRef CAS; (b) W.-H. Chiou, G.-H. Lin, C.-C. Hsu, S. J. Chaterpaul and I. Ojima, Org. Lett., 2009, 11, 2659 CrossRef CAS; (c) A. Gonzalez-Gomez, G. Dominguez and J. Perez-Castells, Tetrahedron, 2009, 65, 3378 CrossRef CAS; (d) S. Saha, C. V. R. Reddy and B. Patro, Tetrahedron Lett., 2011, 52, 4014 CrossRef CAS; (e) J. Szawkalo, S. F. Czarnocki, A. Zawadzka, K. Wojtasiewicz, A. Leniewski, J. K. Maurin, Z. Czarnocki and J. Drabowicz, Tetrahedron: Asymmetry, 2007, 18, 406 CrossRef CAS; (f) D. Huang, F. Xu, X. Lin and Y. Wang, Chem.–Eur. J., 2012, 18, 3148 CrossRef CAS; (g) H.-J. Knoelker and S. Agarwal, Synlett, 2004, 1767 CrossRef CAS; (h) F. D. King, J. Heterocycl. Chem., 2007, 44, 1459 CrossRef CAS; (i) V. Singh, S. Hutait and S. Batra, Eur. J. Org. Chem., 2010, 3684 CrossRef CAS; (j) S. M. Allin, S. N. Gaskell, M. R. J. Elsegood and W. P. Martin, Tetrahedron Lett., 2007, 48, 5669 CrossRef CAS; (k) T. Itoh, M. Miyazaki, K. Nagata, M. Yokoya, S. Nakamura and A. Ohsawa, Heterocycles, 2002, 58, 115 CrossRef CAS.
- (a) M. Chbani and M. Pais, J. Nat. Prod., 1993, 56, 99 CrossRef CAS; (b) B. Hoefgen, M. Decker, P. Mohr, A. M. Schramm, S. A. F. Rostom, H. El-Subbagh, P. M. Schweikert, D. R. Rudolf, M. U. Kassack and J. Lehmann, J. Med. Chem., 2006, 49, 760 CrossRef CAS; (c) S. M. Allin, C. I. Thomas, J. E. Allard, K. Doyle and M. R. J. Elsegood, Tetrahedron Lett., 2004, 45, 7103 CrossRef CAS; (d) S. M. Allin, C. I. Thomas, J. E. Allard, K. Doyle and M. R. J. Elsegood, Eur. J. Org. Chem., 2005, 4179 CrossRef CAS; (e) L. Evanno, J. Ormala and P. M. Pihko, Chem.–Eur. J., 2009, 15, 12963 CrossRef CAS; (f) K. Diker, K. E. Biach, M. D. De Maindreville and J. Levy, J. Nat. Prod., 1997, 60, 791 CrossRef CAS; (g) T. Itoh, M. Miyazaki, K. Nagata, S. Nakamura and A. Ohsawa, Heterocycles, 2004, 63, 655 CrossRef CAS; (h) M.-Y. Chang and S.-T. Chen, Heterocycles, 2003, 60, 99 CrossRef CAS; (i) N. Okada, K. Misawa, M. Kitajima and H. Takayama, Heterocycles, 2007, 74, 461 CrossRef CAS; (j) E. Cheng, J. Botzem, M. J. Wanner, B. E. A. Burm and G.-J. Koomen, Tetrahedron, 1996, 52, 6725 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, NMR spectra for new compounds. CCDC & X-ray crystallography details for compounds 2, 2e, 2h, 2o and 2q. CCDC reference numbers 876034–876038. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra21734a |
| This journal is © The Royal Society of Chemistry 2012 |