Copper sensing with a prion protein modified nanopipette†
Paolo
Actis‡
a,
Alex
McDonald‡
b,
David
Beeler
b,
Boaz
Vilozny
a,
Glenn
Millhauser
b and
Nader
Pourmand
a
aDepartment of Biomolecular Engineering, University of California, Santa Cruz, 1156 High Street, Santa Cruz, CA 95064, USA
bDepartment of Chemistry and Biochemistry, University of California, Santa Cruz, 1156 High Street, Santa Cruz, CA 95064, USA
First published on 26th September 2012
Abstract
Protein–metal interactions determine and regulate many biological functions. Nanopipettes functionalized with peptide moieties can be used as sensors for metal ions in solution.
Solid-state nanopores are a class of nanomaterials that have been successfully applied to the study and detection of numerous biomolecular interactions.1,2 A nanopore can be simply described as a nanometer-sized opening, separating two chambers filled with electrolyte solutions. When a voltage is applied across a nanopore, an ionic current is established, with a magnitude that depends on the size, geometry, and charge of the nanopore opening. Biomolecular analysis can be performed when analytes translocate through the nanopore, giving rise to temporary blockades of the ionic current which are characteristic of the size and charge of the analyte being studied. This configuration (called resistive pulse sensing) enables the analysis of single molecules, but lacks specificity, as biomolecules of similar size and charge cannot be discriminated. An alternative experimental configuration relies on the immobilization of a receptor on the nanopore surface. The interaction of the receptor with its target analyte (if present in the solution) causes a permanent current blockade. Our group pioneered the application of a particular class of nanopores, called nanopipettes, to the study of proteins,3,4 toxins5 and metal ions.6,7 In a nanopipette, the nanopore is situated at the tip of a very sharp quartz needle, which can be easily fabricated at the bench using a laser puller.
In this paper we describe the development of functionalized nanopipettes (Fig. 1a and b) to study peptide–metal interactions. As a model system, we take advantage of the unique metal-binding properties of the prion protein (PrP). This protein is responsible for a class of infectious neurodegenerative diseases termed transmissible spongiform encephalopathies (TSEs).8,9 TSEs are spread via the misfolding of cellular PrP (PrPC) into the infectious scrapie form, PrPSc, which in the process form PrPSc amyloid fibrils. It is now well established that PrP is a copper binding protein with an ability to take up several Cu2+ equivalents in its flexible N-terminal segment (Fig. 1c). Uptake of copper in this domain drives several physiological processes, including PrPC trafficking and endocytosis.10
The PrP copper binding domain is composed of four tandem repeats of the highly-conserved eight amino-acid sequence PHGGGWGQ, termed the octarepeats.11 The four octarepeats coordinate a single Cu2+ ion at low occupancy with a Kd of 0.12 nM, as determined by electron paramagnetic resonance (EPR). The histidine from each octarepeat forms a square-planar coordination environment around the Cu2+ ion. At higher Cu2+ concentration, however, each octarepeat coordinates its own Cu2+ ion with a Kd of 7–12 μM.12,13 In addition, the octarepeats can weakly coordinate a single Zn2+ ion in a tetrahedral geometry with a Kd of ∼200 μM.14 Herein, we describe the immobilization of the prion peptide on quartz conical nanopipettes to probe its interaction with metal ions in solution, in order to develop an all-electrical Cu2+ sensor amenable to in vivo studies.
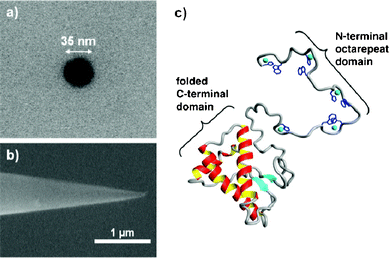 | ||
| Fig. 1 Scanning electron micrographs of the conical nanopipettes used in this study. A) top view showing a 35 nm nanopipette pore and B) side view showing the conical shape. C) Three-dimensional rendering of the prion protein with copper ions included (cyan spheres). | ||
The prion peptide was immobilized by physisorption onto the negatively charged nanopipette walls and the functionalization was followed in real time by chronoamperometry, as described in the Supporting Information.† During immobilization, addition of the prion peptide to the bath solution causes the ionic current to instantaneously drop by ∼35% of its initial value, due to the interaction of the positively charged lysine residues at the N-terminus of the peptide with the negatively charged carboxylic groups of the nanopipette walls (Fig. 2a). The electrostatic immobilization appeared to be stable and the peptide was removed only by treatment with diluted hydrochloric acid (data not shown). The 4-octarepeat is known to bind Cu2+ with micromolar affinity, as demonstrated with mass spectrometry15 and EPR.16 We then added 6 μM Cu2+ to the bath and measured in real-time the evolution of the ionic current (Fig. 2b).The current dropped by 15% in less than a minute, as Cu2+ ions were chelated by histidine moieties of the peptides. A bare nanopipette showed no response to Cu2+ up to mM concentrations. As expected, the binding is reversible by addition of EDTA (ethylenediaminetetraacetic acid) to the bath solution (Fig. S1†). Nanopipette sensors were regenerated up to 6 times with no loss in sensor performance (Fig. 3).
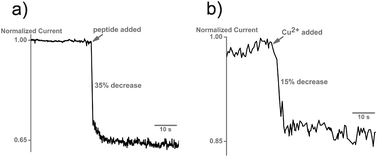 | ||
| Fig. 2 a) Interaction of the prion peptide with the nanopipette surface causes a decrease in the normalized ionic current. 20 μg of the prion peptide was added to the bath. b) Addition of 2 μM Cu2+ causes a further 15% drop in the ionic current, indicative of the interaction of copper with the prion peptide. No change in current is seen when copper is added to an unmodified nanopipette. | ||
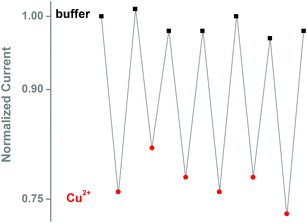 | ||
| Fig. 3 Reversibility of the Cu2+ interaction with the peptide-modified nanopipette. Copper binding was reversed by addition of 1 mM EDTA, and solution was exchanged with fresh buffer. Cu2+ concentration 5 μM. | ||
The current response is concentration dependent (Fig. 4A). We used the functionalized nanopipette to study response of the prion peptide to metal ions. The nanopipette responds linearly to increasing Cu2+ concentrations (Fig. 4A, inset). The modulation of normalized current vs. Cu2+ concentration is analogous to a Langmuir adsorption isotherm. Assuming that copper binding is an equilibrium process with independent binding sites, and the variation of the normalized current is proportional to the number of Cu2+ bound to the nanopipette, the thermodynamic affinity constant K for Cu2+ binding can be estimated using the following equation:
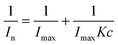 | (1) |
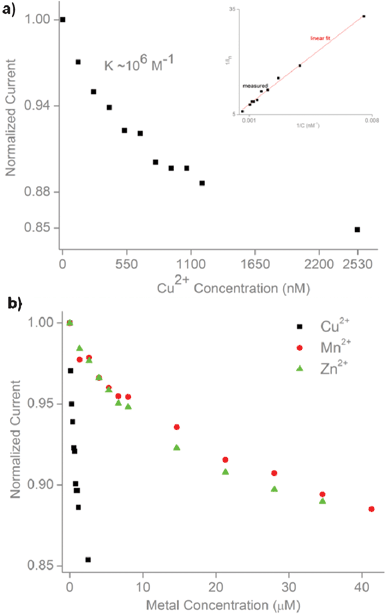 | ||
| Fig. 4 Dose-response and metal screening. a) Dose-response of the peptide-modified nanopipette to increasing concentrations of Cu2+. Inset shows linear fit to the Langmuir isotherm (R2 = 0.99). b) Dose-response to other divalent metals (Mn2+, Zn2+) compared to Cu2+. | ||
Jeremy Lee's group pioneered the application of nanopores to study the interaction of the prion peptide with metals20 and antibodies.21 Lee's group employed a biological nanopore, α-haemolysin, to study metal-induced conformational changes in the prion peptide. They employed resistive-pulse sensing to monitor the change of the translocation rate of prion peptides upon interaction with metal ions. Their approach has the advantage of qualitatively studying peptide–metal interactions at the single-molecule level, but it cannot be used for analytical screening. Our approach, which relies on the immobilization of the prion peptide on the nanopipette walls, allows the quantitative monitoring of peptide–metal interactions and extrapolation of thermodynamic affinity constants. Furthermore, nanopipettes can be easily integrated with nanomanipulators to comprise a scanning ion conductance microscope. This allows positioning of the nanopore sensor over living cells with nanometer resolution.22–24 We propose that, with further improvements, these nanopipette sensors could be used to monitor in real time the extra- and intra-cellular concentrations of metal ions in and around living cells.
In conclusion, electrostatic immobilization of the prion peptide on a conical quartz nanopipette allows the monitoring of peptide-metal interactions. The sensor takes advantage of the unique properties of the PrP octarepeat domain and can be regenerated several times by EDTA treatment without any significant loss of performance, thus demonstrating the stability of the peptide interaction with nanopipette walls. Our findings pave the way for the development of peptide-encoded nanopipette sensors. As shown by Scott Banta's group, peptides selected from phage display can be used as recognition elements in biosensing experiments,25 which expands the applications of nanopipette sensors in biomolecular analysis.
Acknowledgements
We thank Prof. Holger Schmidt and the Keck Center for Nanoscale Optofluidics at UC Santa Cruz for electron microscopy of the nanopores. This work was supported in part by grants from the National Aeronautics and Space Administration Cooperative Agreements NCC9-165 and NNX08BA47A, the National Institutes of Health grants: P01-35HG000205, and GM065790.References
- Z. S. Siwy and S. Howorka, Chem. Soc. Rev., 2010, 39, 1115–1132 RSC.
- U. F. Keyser, J. R. Soc. Interface, 2011, 8, 1369–1378 CrossRef CAS.
- S. Umehara, M. Karhanek, R. W. Davis and N. Pourmand, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4611–4616 CrossRef CAS.
- P. Actis, A. Rogers, J. Nivala, B. Vilozny, R. A. Seger, O. Jejelowo and N. Pourmand, Biosens. Bioelectron., 2011, 26, 4503–4507 CrossRef CAS.
- P. Actis, O. Jejelowo and N. Pourmand, Biosens. Bioelectron., 2010, 26, 333–337 CrossRef CAS.
- P. Actis, B. Vilozny, R. A. Seger, X. Li, O. Jejelowo, M. Rinaudo and N. Pourmand, Langmuir, 2011, 27, 6528–6533 CrossRef CAS.
- B. Vilozny, P. Actis, R. A. Seger, Q. Vallmajo-Martin and N. Pourmand, Anal. Chem., 2011, 83, 6121–6126 CrossRef CAS.
- S. Prusiner, Science, 1991, 252, 1515–1522 CAS.
- S. B. Prusiner, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 13363–13383 CrossRef CAS.
- W. S. S. Perera and N. M. Hooper, Curr. Biol., 2001, 11, 519–523 CrossRef CAS.
- F. Wopfner, G. Weidenhöfer, R. Schneider, A. von Brunn, S. Gilch, T. F. Schwarz, T. Werner and H. M. Schätzl, J. Mol. Biol., 1999, 289, 1163–1178 CrossRef CAS.
- A. P. Garnett and J. H. Viles, J. Biol. Chem., 2003, 278, 6795–6802 CrossRef CAS.
- M. Chattopadhyay, E. D. Walter, D. J. Newell, P. J. Jackson, E. Aronoff-Spencer, J. Peisach, G. J. Gerfen, B. Bennett, W. E. Antholine and G. L. Millhauser, J. Am. Chem. Soc., 2005, 127, 12647–12656 CrossRef CAS.
- E. D. Walter, D. J. Stevens, M. P. Visconte and G. L. Millhauser, J. Am. Chem. Soc., 2007, 129, 15440–15441 CrossRef CAS.
- R. M. Whittal, H. L. Ball, F. E. Cohen, A. L. Burlingame, S. B. Prusiner and M. A. Baldwin, Protein Sci., 2000, 9, 332–343 CrossRef CAS.
- J. S. Hyde, B. Bennett, E. D. Walter, G. L. Millhauser, J. W. Sidabras and E. Antholine William, Biophys. J., 2009, 96, 3354–3362 CrossRef CAS.
- G. L. Millhauser, Annu. Rev. Phys. Chem., 2007, 58, 299–320 CrossRef CAS.
- E. Gaggelli, F. Bernardi, E. Molteni, R. Pogni, D. Valensin, G. Valensin, M. Remelli, M. Luczkowski and H. Kozlowski, J. Am. Chem. Soc., 2004, 127, 996–1006 CrossRef.
- S. P. Leach, M. D. Salman and D. Hamar, Anim. Health Res. Rev., 2006, 7, 97–105 CrossRef.
- R. I. Stefureac, C. A. Madampage, O. Andrievskaia and J. S. Lee, Biochem. Cell Biol., 2010, 88, 347–358 CrossRef CAS.
- C. A. Madampage, O. Andrievskaia and J. S. Lee, Anal. Biochem., 2010, 396, 36–41 CrossRef CAS.
- R. Adam Seger, P. Actis, C. Penfold, M. Maalouf, B. Vilozny and N. Pourmand, Nanoscale, 2012, 4, 5843–5846 RSC.
- C.-C. Chen, Y. Zhou and L. A. Baker, Annu. Rev. Anal. Chem., 2012, 5, 207–228 CrossRef CAS.
- P. Novak, C. Li, A. I. Shevchuk, R. Stepanyan, M. Caldwell, S. Hughes, T. G. Smart, J. Gorelik, V. P. Ostanin, M. J. Lab, G. W. J. Moss, G. I. Frolenkov, D. Klenerman and Y. E. Korchev, Nat. Methods, 2009, 6, 279–281 CrossRef CAS.
- J. Wu, D. M. Cropek, A. C. West and S. Banta, Anal. Chem., 2010, 82, 8235–8243 CrossRef CAS.
Footnotes |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21730a |
| ‡ These authors contributed equally to the work |
| This journal is © The Royal Society of Chemistry 2012 |