Fluorescent detection of silver ions in water with organic nano-aggregates†
Joydev
Hatai
,
Suman
Pal
and
Subhajit
Bandyopadhyay
*
Indian Institute of Science Education and Research (IISER) – Kolkata, BCKV Main Campus PO, Mohanpur, Nadia, WB 741252, India. E-mail: sb1@iiserkol.ac.in
First published on 4th October 2012
Abstract
A novel chemosensor, that has a bis-thiocarbamate scaffold appended to hydrophobic naphthoyl groups, displays fluorescence enhancement in the presence of Ag+ ions. The sensor behaves differently in aqueous and methanolic environments. In the aqueous media it forms organic nano-aggregates, which undergo further aggregation and grow in size in the presence of Ag+ ions. The detection limit of the sensor is 11 ppb for Ag+ ions, which is far within the US-EPA guidelines for drinking water.
Introduction
Silver is one of the persistent ecological contaminants for surface water. Silver salts are commonly used in topical ointments for burn patients because of their antimicrobial properties.1 The overuse of medication containing silver salts, however, results in accumulation of silver in liver tissues and its adverse effect on patients has caused concern.2 The burgeoning use of silver nanoparticles, popularly called nanosilver, in consumer products, particularly in toothpastes, soaps and deodorants as antimicrobial agents, is a matter of grave concern.3 Leaching of silver ions from nanoparticle-containing products has been reported.4 Detection of silver ions in water and biological samples is important.Although analytical methods, such as atomic absorption spectrometry (AAS), inductively coupled plasma-mass spectrometry (ICPMS), inductively coupled plasma-atomic emission spectrophotometry (ICP-AES) and electrochemical techniques, such as ion-selective potentiometry and anodic stripping voltammetry have unparalleled sensitivity,5 fluorescence methods offer a simple alternative for detection and quantification of ions and molecules.6 Although a large number of fluorescence sensors have been reported for Cu2+, Zn2+ and Hg2+, reports of Ag+ detection with fluorescence techniques are relatively few in the literature.7
We have recently reported the differential detection of Hg2+, Pb2+ and Zn2+ using a single chemosensor by tuning of solvents.8 Here, we report a fluorescent sensor bearing two thiocarbamate functionalities that displays high selectivity for Ag+ ions, both in water and in non-aqueous media. Heavy metal ions, such as Ag+ and Hg2+ ions, being “soft” cations according to Pearson's classification9 prefer “soft donors” for complexation. Therefore, in designing the chemosensor for heavy metal ions, soft sulphur donor centres were introduced in the receptor as thiocarbamate moieties. The naphthoyl fluorophore is not an ideal fluorophore for bioimaging since both its excitation and emission occurs in the UV range. However, for in vitro detection that does not pose any serious problems.10 In this case, the naphthoyl unit was introduced in the sensor molecule as a hydrophobic fluorophore. The sensor in methanolic solution displays a ratiometric behaviour in its absorption spectra with addition of Ag+. Under the same conditions in the fluorescence mode, it shows a nine-fold enhancement of the fluorescence intensity (quantum yield, φ from 0.038 to 0.48) and a negligible shift in the emission maxima. In aqueous media, the lipophilic ligand forms nano-aggregates, as revealed by dynamic light scattering (DLS) studies. The nano-aggregates underwent further aggregation resulting in larger aggregates in the presence of Ag+. The nano-aggregates in water displayed a 10 nm red shift compared to the non-aggregated ligand in methanol. On addition of silver ions the emission maxima further shifted by another 15 nm with a six-fold enhancement of the fluorescence intensity (φ from 0.028 to 0.21).
Results and discussion
The bipodal chemosensor 1 consists of a two dithiocarbamate moieties attached to naphthoyl units via ethylenediamine spacers. The key steps for the synthesis of 1 is shown in Scheme 1. A naphthoyl–ethylenediamine intermediate 2 was synthesized by coupling naphthoyl chloride with Boc protected ethylenediamine 3 in 85% yield. Deprotection of the Boc group, followed by treatment with carbon disulfide and the bromomethyl compound 411 afforded the thiocarbamate compound 1 in a single step, which on chromatography yielded 80% of the product (see Experimental section and Scheme 1). Compound 1 was characterized by 1D and 2D NMR and other spectroscopic and spectrometric methods.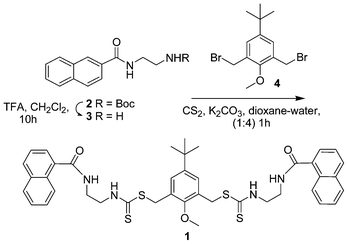 | ||
| Scheme 1 Synthesis of chemosensor 1. | ||
To investigate the properties of 1 as a chemosensor, UV-vis and fluorescence experiments were initially carried out using perchlorate salts of Na+, K+, Cr3+, Mn2+, Fe2+, Fe3+, Hg2+, Co2+, Ni2+, Cu2+, Zn2+, Pb2+ and AgNO3 with compound 1 in CH3OH/DMSO (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). Out of the ions mentioned above, a visible change from colourless to yellow was observed only with Ag+ (Fig. 5C). Sensor 1 in the absence of any metal showed a band at 272 nm and a weaker band at 325 nm (Fig. 1).
:1, v/v). Out of the ions mentioned above, a visible change from colourless to yellow was observed only with Ag+ (Fig. 5C). Sensor 1 in the absence of any metal showed a band at 272 nm and a weaker band at 325 nm (Fig. 1).
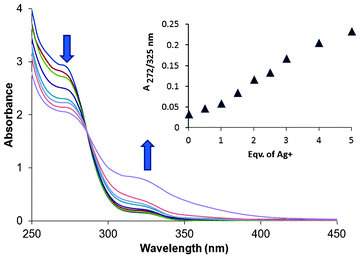 | ||
| Fig. 1 Change in the absorption spectra of 1 (100 μM) upon addition of AgNO3 (0–500 μM). The purple trace tailing to ca. 400 nm was obtained after mild heating of the 500 μM sample of Ag+ with 1 (100 μM) (see text). | ||
With addition of silver salt, the intensity of the band at 272 nm gradually diminished, whereas the one at 325 nm increased. The tail of the spectra, which ended at 355 nm in the absence of Ag+, enhanced significantly in intensity and underwent a red shift to ca. 400 nm on addition of silver salts, displaying a ratiometric behaviour. The colour change is possibly a result of the charge transfer from the sulphur donors of the ligand to the silver ion.12 There was no significant change in the absorption spectra with any of the other metal ions. On mild heating, the yellow colour of the solution containing 1 and Ag+ became more intense. At basic pH no colour change of the ligand solution was observed, eliminating the possibility that the colour change was a result of deprotonation of ligand 1.
The fluorescence behaviour of 1 with Ag+ was investigated under different proportions of MeOH/DMSO/H2O (Fig. 2B). Upon excitation of the naphthoyl fluorophore at 290 nm, the fluorescence intensity at 360 nm was enhanced ninefold (increase in quantum yield, φ from 0.038 to 0.48) in MeOH/DMSO (99:1, v/v) with a negligible 2 nm shift in the fluorescence peak (Fig. 2A). Most of the other ions showed either a negligible change or a small quenching effect with 1 (Fig. 6A). The increase in the fluorescence intensity with a negligible shift in the emission maxima is characteristic for systems where the inhibition of the photo-induced electron transfer (PET) mechanism takes place.13
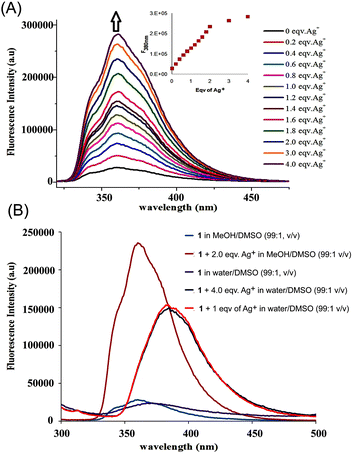 | ||
| Fig. 2 (A) Fluorescence enhancement of 1 with addition of AgNO3 (0–4 equiv.) in CH3OH/DMSO (99:1, v/v), (λex = 290 nm). The inset shows a decrease in the rate of fluorescence enhancement after the addition of 2 equiv. of Ag+. (B) Fluorescence intensity of 1 with addition of AgNO3 (0–4 equiv.) in CH3OH/DMSO (99:1, v/v) and H2O/DMSO (99:1, v/v), (λex = 290 nm). | ||
To understand the basis of the fluorescence enhancement, the orbital maps of the HOMO and the LUMO were generated from the energy-minimized structure of receptor 1 (Fig. 3) with semi-empirical ZINDO calculations. The contours suggested that the highest occupied levels were largely centred on the electron rich thiocarbamate moieties, while the lowest unoccupied level was naphthoyl centred. Thus, the PET process involved the transition of the electron from a sulphur to the naphthalene unit. This was further supported by the fact that at lower pH values there was no significant enhancement of the fluorescence intensity of the sensor 1, which is typical for a PET system with a S-centred donor.
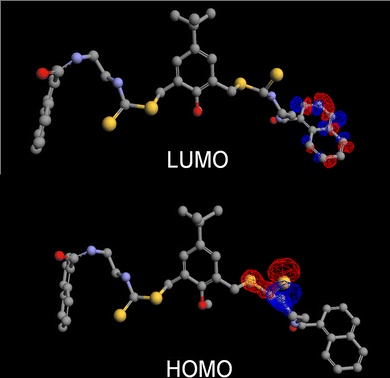 | ||
| Fig. 3 The orbital map of the HOMO and LUMO levels of chemosensor 1. | ||
Interestingly, at first, when the fluorescence readings were recorded immediately after the addition of the metal ions, we failed to detect any Hg2+ ions. However, when the fluorescence spectra were recorded 15 min after the addition of the salts, 1 was found to elicit a fluorescence response with Hg2+ as well. It is also noteworthy that there was no colour change on addition of Hg2+ to 1 (Fig. 5A; also see Fig. S1†). The affinity of the sensor for the soft Ag+ and Hg2+ metal ions is because of the presence of the soft sulphur donor centres of 1.
To investigate the binding properties of Ag+ with 1 in CH3OH/DMSO (99:1, v/v), fluorescence titration was carried out with a series of samples obtained by varying the amount of Ag+ added to a constant amount of 1 (Fig. 2A). Upon addition of up to two equivalents of the metal ion, steady growth of the fluorescence intensity was observed from the series of the spectra. After two equivalents, the rate of increase of the fluorescence intensity became substantially lower (inset: Fig. 2A), indicating the formation of a 2:1 metal–ligand complex of a silver ion with sensor 1. Indeed, a Job's plot experiment (Fig. 5A) at a total concentration of 8 μM revealed a 2:1 stoichiometry.
This was separately confirmed when analysis of the fluorescence data was performed using the methods of Lehrer and Chipman (eqn (1))14 for the calculation of the binding constant of the (Ag+n·1) complex:
| ln[(F − F0)/(F∞ − F)] = nln [Ag+] + nln (Kasscn) | (1) |
In eqn (1), n refers to the number of silver ions associating with each molecule of 1, Kasscn refers to the association constant, F0, F and F∞ refers to the fluorescence intensities of the solutions of chemosensor 1 alone, 1 in the presence of Ag+ at any concentration and at high concentration (4 equivalents) of Ag+ ion, respectively. The plot of ln[(F − F0)/(F∞ − F)] against ln [Ag+] gave a value of 1.94 (Fig. S5†), which is well in agreement with the 2:1 ratio obtained directly from the fluorescence titration experiments. From the intercept of the best fit straight line, an association constant of 8.0 (±0.6) × 105 M−2 was obtained (the error was obtained from triplicate experiments). A peak at m/z 1034 in electrospray ionization mass spectrometry corresponding to two Ag+ ions associating with one molecule of 1 under these conditions further supported the binding stoichiometry (Fig. S4†). Thus, a putative model for the binding in methanol is shown in Fig. 4.
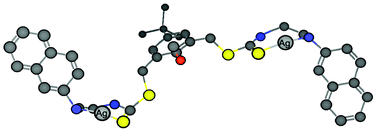 | ||
| Fig. 4 A proposed model for the binding of Ag+ with chemosensor 1 in a 2:1 ratio. | ||
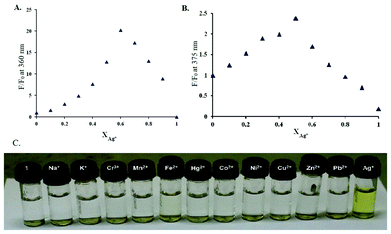 | ||
| Fig. 5 (A) The Job's plot of CH3OH/DMSO (99:1, v/v). (B) The Job's plot of H2O/DMSO (99:1, v/v). (C) A photograph of sensor 1 (100 μM) containing 400 μM of metal ions (200 μM for Ag+) in HEPES buffer solutions (buffered at pH 7.5) after mild heating (50 °C, 2 min). | ||
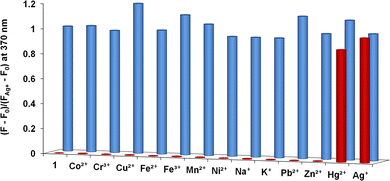 | ||
| Fig. 6 Changes in fluorescence intensity of the chemosensor 1 (10 μM) in CH3OH/DMSO (99:1, v/v) upon addition of 50 μM of various metal perchlorates (red bar). The competitive selectivity of 1 (10 μM) towards Ag+ (10 μM) in the presence of different metal ions (50 μM) in H2O/DMSO (99:1, v/v) (blue bar). | ||
In aqueous solution (H2O/DMSO, 99:1, v/v), the fluorescence intensity of 1 (10 μM) without any metal ion underwent a red shift by 10 nm (Fig. 2B). Upon addition of one equivalent of Ag+, the enhancement of the fluorescence band was only sixfold (φ = 0.028 to 0.21). In addition, under aqueous conditions, a further 15 nm red shift of the fluorescence band was observed. Here, the saturation of the fluorescence intensity was observed with addition of only one equivalent of the Ag+ ion (Fig. 2B). With other metal ions the change was negligible. Again, with Hg2+ the fluorescence response was slow compared to the Ag+ ion.
Under aqueous conditions (H2O/DMSO, 99:1, v/v, HEPES buffer, pH 7.5) the 1:1 association of Ag+ with 1 was confirmed by the Job's plot experiment at a total concentration of 8 μM (Fig. 5B). The binding constant was found to be 1.37 (±0.4) × 106 M−1 (Fig. S6†). The m/z peak at 874 in ESI-MS in this case, also supported the 1:1 association of Ag+ with 1 (Fig. S3†).
The effect of Ag+ on 1 was also studied with 1H NMR spectroscopy in CD3OD and also in d6-DMSO/D2O (2:3, v/v).15 The 1H NMR spectra were recorded for solutions containing 1 and different equivalents of Ag+. The effect of the metal binding was manifested with a change in chemical shifts for several protons of compound 1 (Fig. 7A and B). In CD3OD, there was hardly any change for the resonance of the naphthalene protons, whereas in DMSO/D2O there was significant change in the aromatic protons, especially for H-1 of the naphthalene ring, possibly because of the fact that the two naphthoyl bump into each other because of the close proximity after the binding of Ag+ (see Fig. 9). This is further supported by the fact that the CH2 protons of ethylenediamine moieties next to the carboxynaphthyl group (protons c) underwent a larger upfield shift, most likely because of the change in the conformation of the ligand, where the CH2 protons move towards the shielding zone of the central aryl ring. This explanation has to be taken with caution since the NMR experiments were performed in a DMSO/D2O mixture, whereas the proposed model relies on the energy optimized structure of a single molecule in water.
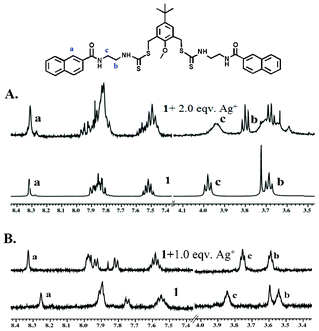 | ||
| Fig. 7 Partial 1H-NMR spectra changes of chemosensor 1 (A) in CD3OD and (B) in DMSO-d6/D2O (2:3, v/v) upon addition of AgNO3. The low quality of the spectra in DMSO/D2O is because of the low solubility of the samples at high concentration. | ||
However, in CD3OD the CH2 protons of ethylenediamine moieties next to the thiocarbamate group underwent a larger downfield shift, indicating that the binding of the silver ions in this case was proximal to the thiocarbamate moiety.
To evaluate the practical application of the chemosensor in analytical chemistry, competition experiments were also carried out to investigate the effect of other coexisting cations along with Ag+ in the presence of the chemosensor. Fluorescence spectra of solutions containing five equivalents of Na+, K+, Cr3+, Mn2+, Fe2+, Fe3+, Hg2+, Co2+, Ni2+, Cu2+, Zn2+ and Pb2+ (50 μM) added to 10 μM of 1 followed by addition of Ag+ (10 μM) were recorded. The results are shown in Fig. 6B. These results clearly demonstrate that the fluorescence response of 1 is not affected by the presence of the other metal ions.
The delayed response of Hg2+ with 1 prompted us to study a time-course of the fluorescence response of both Ag+ and Hg2+ with 1 (Fig. 8). The time-course clearly demonstrates the difference in behaviour of the two metal ions. Based on the time-course experiment, the two ions can be differentiated using UV-vis spectroscopy where Hg2+ does not display any change (Fig. S1†).
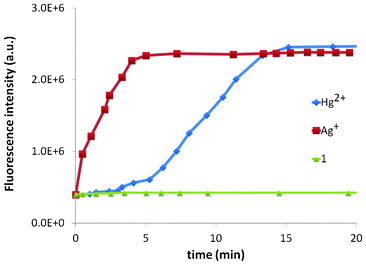 | ||
| Fig. 8 Time-course analysis of the fluorescence response of both Ag+ and Hg2+ with 1. | ||
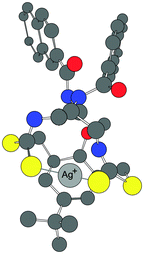 | ||
| Fig. 9 The proposed model of 1:1 binding of the receptor 1 with Ag+ in water. | ||
The analytical detection limit of chemosensor 1 for the detection of Ag+ determined by standard methods16 was found to be as low as 1.0 × 10−7 M, i.e., 11 ppb (Fig. S7†). The maximum contaminant level, as set by the United States EPA, for silver ions is 0.1 ppm,17 which is far within the detection limit of our sensor.
The calculated log P value18 of 9.42 for 1 indicated a high lipophilic character of the ligand. The proposed binding of receptor 1 with a Ag+ ion is shown in Fig. 9. The model is based on the Job's plot experiment in water (Fig. 5B), the ESI-MS (Fig. S3†) supporting the 1:1 binding and the energy optimized structure, which suggests that the two sulphur atoms of both the thiocarbamate moieties bind to the Ag+ ion. In addition, there can be a charge–π interaction between the electron rich central aryl ring and the metal ion. Thus, in the complex, the naphthoyl units provide lipophilicity, whereas the other end of the molecule, on binding to Ag+, provides a polar nature.
To investigate whether the difference in the fluorescence behaviour of 1 in methanol and water was a result of aggregation of the Ag+–ligand complex in water, dynamic light scattering experiments were performed.19 DLS measurements were recorded with 10 μM optically clear solutions of 1 in both methanol and water in the presence and absence of the metal ion. The DLS data of the methanolic solution was essentially the same as that of the background indicating the absence of any aggregation. However, under aqueous conditions (H2O/DMSO, 99:1, v/v) formation of organic nanoparticles with an average hydrodynamic diameter of 94 nm (weighted standard deviation = ±3 nm, polydispersity index PDI = 0.140) was obtained from the particle size distribution analysis of the light scattering data (Fig. 10). Interestingly, larger particles with an average diameter of 196 (±7) nm (PDI = 0.231) were obtained on addition of Ag+, indicating further agglomeration of the particles in the presence of the metal ion. Thus, the enhancement of the fluorescence signal upon addition of Ag+ may be because of two factors: first, the binding of the metal ion to the receptor molecule inhibits the PET process. Second, the aggregation of the [1·Ag+]complex shields at least some of the hydrophobic fluorophore units from water and reduces the quenching effect.
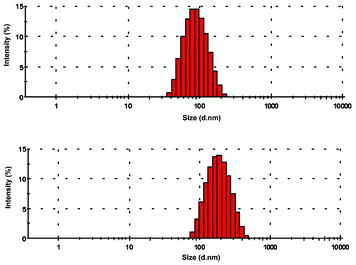 | ||
| Fig. 10 Size distribution analysis of the organic nano-aggregates of 1 using dynamic light scattering experiments in water. The red bars represent the relative population of the corresponding particle size given in an exponential scale in the x-axis. | ||
The pH dependence of the chemosensor was investigated. The fluorescence spectra of the chemosensor in the presence of a silver ion were recorded for a series of samples from pH 3.9 to 11 (Fig. 11). At low pH, the fluorescence response of the chemosensor to Ag+ was low. A good fluorescence response was obtained between pH 7 to 10. The error bars are given from triplicate experiments and the maximum error was 2.8%. At pH higher than 11, the solution became murky and the sensitivity of the sensor went down, probably because of the formation of silver hydroxide. The reversibility of the fluorescence response with the sensor in the presence of Ag+ was observed on addition of sodium chloride.
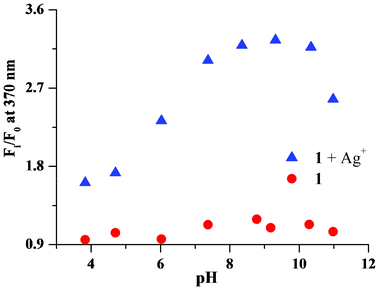 | ||
| Fig. 11 Change in the fluorescence intensity (at 370 nm) of chemosensor 1 in the presence of silver ions at different pH. | ||
Conclusions
In conclusion, fluorescence sensor 1 displays a remarkable selectivity and a 11 ppb sensitivity for Ag+ ions at a physiologically relevant pH range in aqueous, as well as in organic media. In addition, it offers a ratiometric detection in the absorption mode. Sensor 1 also shows a fluorescence response with Hg2+. The sensor differentiates between the two metal ions (Ag+ and Hg2+) on the basis of time-course fluorescence experiments and also with UV-vis spectroscopy, besides the obvious visual change that occurs only with Ag+. In the aqueous media, 1 forms organic nanoparticles (hydrodynamic diameter, dH = 94 ± 3 nm) and the fluorescence enhancement on addition of Ag+ is attributed to the complexation of Ag+ with 1 and subsequent aggregation of the nanoparticles to form a nano-aggregate ensemble with (dH = 196 ± 7 nm). Variation of the fluorophore on this scaffold is under way for NIR detection and imaging of biologically relevant analytes.Experimental section
General information
The compound’s structure was determined by nuclear magnetic resonance (NMR). 1H and 13C NMR spectra were recorded at 400 MHz (Jeol) and 500 MHz (Bruker), respectively. Chemical shifts are reported in δ values relative to an internal reference of tetramethylsilane (TMS) for 1H NMR and the solvent peak in 13C NMR, except where noted. Peak splitting patterns in the NMR are reported as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet and br, broad. IR data were obtained with a Bruker-Optics-Alpha-T spectrophotometer. UV spectra were recorded with a Hitachi U-4100. Fluorescence measurements were carried out using a Fluoromax-3 (Horiba Jobin Yvon). Mass data were obtained from an Acquity TM ultra performance LC. pH data were recorded using a Sartorius Basic Meter PB-11. Reactions were monitored by thin layer chromatography using Merck plates (TLC Silica Gel 60 F254). Developed TLC plates were visualized with UV light (254 nm). Silica gel (100–200 mesh, Merck) was used for column chromatography. Yields indicate the chromatographically and spectroscopically pure (>95%) compounds, except as otherwise indicated.Fluorescence quantum yield was determined using 2-naphthoic acid as a standard. All quantum yields were measured with solutions containing 0.1, 0.5, 1.0 and 5.0 μM of each of the samples. All samples were deaerated with nitrogen gas prior to the measurements. Each of the experiments was repeated in triplicate to obtain a maximum of 10% error in the data.
Procedure for metal ion sensing
For absorption and emission spectra, stock solutions of metal perchlorate salts (1.0 mM) and free chemosensor 1 (10 μM) were prepared in DMSO/CH3OH (1:99, v/v) and DMSO/H2O (1:99) in the presence of HEPES buffer (1 mM). All experiments were carried out with 10 μM solutions of 1 in DMSO/MeOH, DMSO/H2O mixtures and various concentrations of metal perchlorate in methanol and water, respectively. Excitation was performed at 290 nm with all excitation and emission slit widths at 5.0 nm unless otherwise indicated.
Dynamic light scattering studies
The particle size of the aggregates was measured by dynamic light scattering (DLS) experiments on a Malvern Zetasizer Nano ZS instrument equipped with a 4.0 mW He–Ne laser operating at a wavelength of 633 nm. The samples and the background were measured at room temperature at a scattering angle of 173°.Synthesis
:1, v/v) yielded 2 (0.58 g, 89%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.27 (s, 1H, –ArH), 7.80 (m, 4H, –ArH), 7.45 (m, 2H, –ArH), 7.35 (br, 2H, –NH), 3.54 (td, J = 5.5 Hz, 2H, –CH2NH), 3.36 (t, J = 5.5 Hz, 2H, –CH2NHBoc), 1.36 (s, 9H, –Boc).
:4) (5 mL) was stirred for 15 min at 0 °C. Carbon disulfide (1.74 g, 1.4 mL, 23.22 mmol) was added to this reaction mixture and continued to be stirred for another 10 min. The dibromomethyl compound 4 (0.20 g, 0.57 mmol) was dissolved in dioxane (3 mL) and was added drop wise over a period of (10–15) minutes. The obtained solution warmed up to room temperature and was stirred for 30 min. After TLC showed complete conversion, the solution was concentrated under reduced pressure. The solid mass obtained was dissolved in methylene chloride (15 mL) and washed with water. The organic layer was dried over anhydrous Na2SO4 and volatiles were removed under reduced pressure. The compound on chromatography (hexane/ethyl acetate, 1:4, v/v) yielded (0.35 g, 80%) as a white fluffy solid, m.p. 99–100 °C; 1H NMR (500 MHz, CDCl3) δ 9.24 (t, J = 5.0 Hz, 2H, –CONHCH2), 8.20 (s, 2H, ArH), 7.77 (t, 2H, J = 5 Hz, –NHCS), 7.72 (d, J = 8.0 Hz, 4H,ArH), 7.66 (t, J = 7.5 Hz, 4H, ArH), 7.47 (t, J = 7.5 Hz, 2H, ArH), 7.39 (t, J = 7.5 Hz, 2H,ArH), 7.26 (s, 2H, ArH), 4.31 (s, 4H, –ArCH2), 4.05 (td, J = 5 Hz, 4H, –CONHCH2), 3.80 (td, J = 5.0 Hz, 4H, –CH2NHCS), 3.76 (s, 3H, –OCH3). 13C NMR (125 MHz, CDCl3): 197.97, 168.77, 152.57, 148.85, 134.61, 132.32, 130.67, 128.84, 128.73, 128.29, 127.76, 127.65, 127.51, 126.59, 123.39, 62.98, 46.59, 40.32, 34.62,34.35, 31.09. UV-vis (MeOH; nm) 290, 360. FT–IR (KBr, cm−1): 3230, 2959, 1642, 1625, 1528, 1504 1483, 1304, 1235, 1173, 973, 777, 760. ESI-MS calcd for C41 H44N4O3 S4Na+ 791.2, found 791.1. HRMS calcd for C41H44N4O3S4 768.2294; found: 768.2292.
Acknowledgements
The generous help of Mr. Santu Sarkar with the DLS studies is gratefully acknowledged. We thank the CSIR for a fellowship to JH, IISER for facilities and DST (SR/S1/OC-26/2010) for a research grant. This paper is dedicated to a wonderful mentor and an inspiring teacher Professor Ronald Breslow.References
- Review on effect of silver on burn wound infection control and healing: S. Atiyeh, B. M. Costagliola, S. N. Hayek and S. A. Dibo, Burns, 2007, 33, 139–148 CrossRef.
- (a) I. Chopra, J. Antimicrob. Chemother., 2007, 59(4), 587–590 CrossRef CAS; (b) M. Kazuyuki, H. Nobuo, K. Takatoshi, K. Yuriko, H. Osamu, I. Yashihisa and S. Kiyoko, Clin. Chem., 2001, 47, 763–766 Search PubMed.
- (a) Advisory Committee on Hazardous Substances. Report on Nanosilver, Department for Environment, Food and Rural Affairs: London, 2009; p 7 Search PubMed; (b) R. Kessler, Environ. Health Perspect., 2011, 119(3), 121–125, DOI:10.1289/ehp.119-a120.
- B. M. Troy and P. Westerhoff, Environ. Sci. Technol., 2008, 42, 4133–4139 CrossRef.
- K. A. Anderson, Mercury Analysis in Environmental Samples by Cold Vapor Techniques. In Encyclopedia of Analytical Chemistry, R. A. Meyes, Ed., Wiley: New York, 2006 Search PubMed.
- (a) B. Valeur, J. Bourson, J. Pouget and A. W. Czarnik, Fluorescent Chemosensors for Ion and Molecule Recognition; ACS Symposium Series 538; American Chemical Society: Washington, DC, 1993 Search PubMed; (b) A. W. Czarnik, Acc. Chem. Res., 1994, 27, 302 CrossRef CAS; (c) B. Wang and E. J. Anslyn, Chemosensors: Principles, Strategies and Applications, John Wiley and Sons, New York, 2011 Search PubMed; (d) C. D. Geddes and J. R Lakowicz, Advanced Concepts in Fluorescence Spectroscopy: Small Molecule Sensing: Topics in Fluorescence Spectroscopy: Vol 9; Springer: New York, 2005 Search PubMed.
- (a) W. Fang, R. Nandhakumar, K. M. Kim and J. Yoon, Inorg. Chem., 2011, 50, 2240–2245 CrossRef; (b) D.-H. Li, J.-S. Shen, N. Chen, Y.-B. Ruana and Y.-B. Jiang, Chem. Commun., 2011, 47, 5900–5902 RSC; (c) S. Huang, S. He, Y. Lu, F. Wei, X. Zeng and L. Zhaoab, Chem. Commun., 2011, 47, 2408–2410 RSC; (d) M. Kumar, R. Kumar and V. Bhalla, Org. Lett., 2011, 13, 366–369 CrossRef CAS; (e) H.-H. Wang, L. Xue, Y.-Y. Qian and H. Jiang, Org. Lett., 2010, 12, 292 CrossRef CAS; (f) S. S. Tan, Y. N. Teo and E. T. Kool, Org. Lett., 2010, 12, 4820–4823 CrossRef CAS; (g) Y.-H. Lina and W.-L. Tseng, Chem. Commun., 2009, 6619–6621 RSC; (h) A. Chatterjee, M. Santra, N. Won, S. Kim, K. J. Kim, B. S. Kim and H. K. Ahn, J. Am. Chem. Soc., 2009, 131, 2040 CrossRef CAS; (i) R. Joseph, B. Ramanujam, A. Acharya and C. P. Rao, J. Org. Chem., 2009, 74, 8181 CrossRef CAS; (j) S. Iyoshi, M. Taki and Y. Yamamoto, Inorg. Chem., 2008, 47, 3946–3948 CrossRef CAS; (k) L. Liu, D. Zhang, G. Zhang, J. Xiang and D. Zhu, Org. Lett., 2008, 10, 2271 CrossRef CAS; (l) E. U. Akkaya and A. Coskun, J. Am. Chem. Soc., 2005, 127, 10464 CrossRef.
- J. Hatai, S. Pal, G. P. Jose, T. Sengupta and S. Bandyopadhyay, RSC Adv., 2012, 2, 7033–7036 RSC.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- (a) A. Banerjee, A. Sahana, S. Guha, S. Lohar, I. Hauli, S. K. Mukhopadhyay, J. S. Matalobos and D. Das, Inorg. Chem., 2012, 51, 5699–5704 CrossRef CAS; (b) A. K. Mandal, M. Suresh, P. Das, E. Suresh, M. Baidya, S. K. Ghosh and A. Das, Org. Lett., 2012, 14, 2980–2983 CrossRef CAS; (c) X. He, Q. Zhang, W. Wang, L. Lin, X. Liu and X. Feng, Org. Lett., 2011, 13, 804–807 CrossRef CAS; (d) D-H. Li, J.-S. Shen, N. Chen, Y.-B. Ruana and Y.-B. Jiang, Chem. Commun., 2011, 47, 5900–5902 RSC; (e) X-L. Tang, X.-H. Peng, W. Dou, J. Mao, J.-R. Zheng, W.-W. Qin, W.-S. Liu, J. Chang and X.-J. Yao, Org. Lett., 2008, 10, 3653–3657 CrossRef CAS.
- M. Ashram, D. O. Miller, J. N. Bridson and P. E. Georghiou, J. Org. Chem., 1997, 62, 6476 CrossRef CAS.
- (a) M. G. Babashkina, D. A. Safin, M. Bolte and Y. Garcia, Dalton Trans., 2011, 40, 8523–8526 RSC; (b) Y. Li and T. Michinobu, J. Mater. Chem., 2012, 22, 9513 RSC.
- A. P. De Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef CAS.
- (a) S. S. Lehrer and G. D. Fashman, Biochem. Biophys. Res. Commun., 1966, 2, 133–138 CrossRef; (b) D. M. Chipman, V. Grisaro and N. Shanon, J. Biol. Chem., 1967, 242, 4388–4394 CAS; (c) S. Saha, A. Ghosh, P. Mahato, S. Mishra, S. K. Mishra, E. Suresh, S. Das and A. Das, Org. Lett., 2010, 12, 3406–3409 CrossRef CAS.
- The NMR spectra were difficult to obtain in pure D2O because of the aggregation of 1.
- (a) M. Shortreed, R. Kopelman, M. Kuhn and B. Hoyland, Anal. Chem., 1996, 68, 1414–1418 CrossRef CAS; (b) W. Lin, L. Yuan, Z. Cao, Y. Feng and L. Long, Chem. Eur. J., 2009, 15, 5096–5103 CrossRef CAS.
- http://water.epa.gov/drink/contaminants/index.cfm (accessed on July 18, 2012).
- A. Leo, C. Hansch and D. Elkins, Chem. Rev., 1971, 71, 525–616. The log P value has been calculated using MedChem Designer (TM) version 1.0.1.15, Simulations Plus, Inc., 2011 Search PubMed.
- M. T. Morgan, P. Bagchi and C. J. Fahrni, J. Am. Chem. Soc., 2011, 133, 15906–15909 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Detailed characterization of compound 1 along with the intermediates and additional spectroscopic details are provided. See DOI: 10.1039/c2ra21717a |
| This journal is © The Royal Society of Chemistry 2012 |