Superhydrophobic hollow spheres by electrodeposition of fluorinated poly(3,4-ethylenedithiopyrrole)
Herve
Bellanger
,
Thierry
Darmanin
,
Elisabeth
Taffin de Givenchy
and
Frederic
Guittard
*
Université de Nice Sophia-Antipolis & CNRS, Laboratoire de Physique de la Matière Condensée, Equipe Surfaces & Interfaces, Parc Valrose, Nice Cedex 2, 06108, France. E-mail: Frederic.GUITTARD@unice.fr; Fax: (+33)4-92-07-61-56; Tel: (+33)4-92-07-61-59
First published on 3rd October 2012
Abstract
Here, we report for the first time the possibility of obtaining hollow spheres in one pot by electrodeposition of fluorinated poly(3,4-ethylenedithiopyrrole)s. From synthetic pathway to elaborate heterocyclic core (eight steps), we report the synthesis of 2-(2H-[1,4]dithiino[2,3-c]pyrrol-6(3H)-yl)ethanol, also named N-(2-hydroxyethyl)-3,4-ethylenedithiopyrrole, as a new platform molecule to build up conductive materials. From this synthon, fluorinated (F-octyl, F-hexyl and F-butyl) 3,4-ethylenedithiopyrrole derivatives are synthesized and used as monomers for electrodeposition experiments. Their characterization by cyclic voltammetry reveals that the conjugation length is very short due to the presence of two sulfur atoms at the 3- and 4-positions of pyrrole. The films display superhydrophobic properties with relatively low hysteresis and sliding angle. The surface morphology consists of assembly of spherical particles. The mean size of the particles as well as the possibility of forming hollow structures increases as the fluorinated chain length decreases. The use of theoretical nucleation and growth mechanisms models reveals that a progressive three-dimensional growth as well as the relative solubility of the polymers (short polymer chains and short fluorinated tails) seem to be the determining parameters in the formation of hollow structures.
1. Introduction
The research into hollow structures like hollow spheres is very active due to their many potential applications in catalysis, drug delivery, optical devices, sensors, solar cells or batteries.1–12 Many strategies have been used in the literature to produce such structures like the use of hard templates (polymer beads,13 colloidal particles),14 and the use of soft templates (micelles,15 vesicles16 and gas bubbles).17 Many kinds of materials can be obtained such as metals,18 oxides19 or polymers.20 Among them, conducting polymers offer exceptional properties like electronic and optical properties, lightness and ease of functionalization.21The geometrical shape of the hollow spheres is also a very interesting parameter in the elaboration of superhydrophobic surfaces. Indeed, such surfaces required the combination of adequate surface structure and intrinsic hydrophobicity of the polymers.22–26 Sun et al. used polystyrene beads as sacrificial template for the preparation of hollow urchin-like structures of polyaniline.27 After spreading of the solution on glass slides coated with a glue, the surface displayed superhydrophobic properties with reversible switching from superhydrophobic to superhydrophilic by changing the pH and the electrical potential. Another group reported the elaboration of superhydrophobic surfaces formed by hollow dandelion-like and rambutan-like structures obtained through the self-assembly of polyaniline nanofibers.28,29 Superhydrophobic hollow bars were also produced using a self-removing metal-monomeric template, which consisted of a complex between Cd2+ and aniline.30
Conducting polymers can also be deposited on conductive substrates by electrodeposition. If a hydrophobic substituent is grafted on the monomer before polymerization, this process can lead to superhydrophobic surfaces in one step.31–36 The surface morphology and as a consequence the wetting properties of the films are highly dependent on electrochemical parameters and the chemical structure of the monomer.31–36 Various surface morphologies like fibers,33a fibrils,33b needles,33b flower-like structures,34b cauliflower-like structures34 and nanoporous structures31,32 were reported in the literature but to our knowledge this technique was not reported as a method to obtain hollow spheres.
Here, we show that the original fluorinated 3,4-ethylenedithiopyrrole (EDTP), represented in Scheme 1, can be used to electrodeposit, in one step, hollow spheres displaying superhydrophobic properties. Because no functionalized EDTP derivatives were reported in the literature, a synthesis strategy was implemented in order to introduce a hydroxyl substituent and fluorinated chains (Scheme 2). The polymer films were characterized by cyclic voltammetry, contact angle measurement, scanning electron microscopy and optical profilometry. The growth mechanism was also studied using theoretical nucleation and growth mechanism models. All these characterizations were performed to determine the key parameters in the formation of hollow spheres.
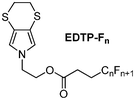 | ||
| Scheme 1 The fluorinated EDTP derivatives studied (EDTP-Fn with n = 4, 6, 8). | ||
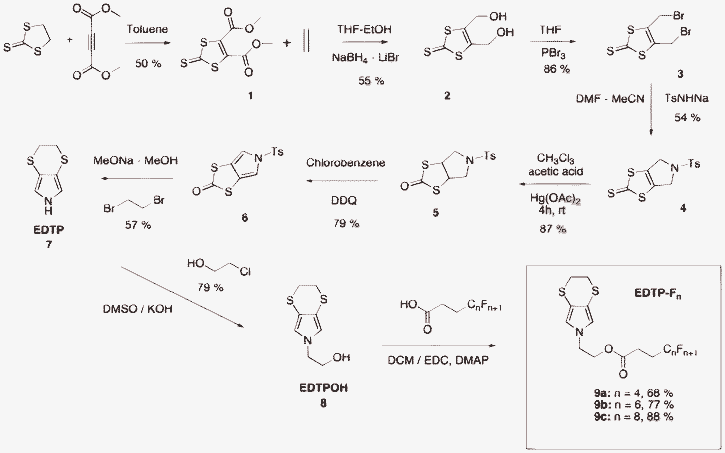 | ||
| Scheme 2 Synthetic route to monomers EDTP-Fn. | ||
2. Experimental
2.1. Monomer synthesis and characterization
3,4-Ethylenedithiopyrrole (EDTP) was synthesized in seven steps.37,38 The original N-(2-hydroxyethyl)-EDTP (EDTPOH) was synthesized using the reaction reported by Oestreich et al. for the preparation of hydroxyalkylpyrrole.39EDTP (3 eq, 220 mg, 1.4 mmol) was added to a suspension of KOH (4 eq, 105 mg, 1.9 mmol) in DMSO (2.5 mL). The reaction mixture was stirred for one hour and cooled in an ice bath before the dropwise addition of a solution of 2-chloroethanol (1 eq, 37 mg, 0.46 mmol) in DMSO (1 mL). After stirring at room temperature for 24 h, water was added and the aqueous phase was extracted using dichloromethane. The combined organic layers were washed with brine and dried over sodium sulfate. Evaporation of the solvent and column chromatography purification (silica gel, cyclohexane/ethyl acetate 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) gave EDTPOH as a yellow oil.
:2) gave EDTPOH as a yellow oil.
Yield 79%; yellow oil; δH(200 MHz, CDCl3) 6.47 (2 H, s), 3.85 (2 H, t, J 4.7), 3.73 (2 H, t, J 4.7), 3.14 (4 H, s); δC(50 MHz, CDCl3): 117.34, 107.09, 62.41, 52.15, 27.09; MS (70 eV): m/z 201 (M+, 100), 170 (C7H8S2+•, 23), 142 (C5H4S2+•, 67); FTIR (KBr): νmax/cm−1 3405, 2913, 1641, 1499, 1316, 1054, 768 cm−1.
Finally, the monomers EDTP-Fn were successfully obtained using 3-F-alkylpropanoic acids, with N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) and 4-dimethylaminopyridine (DMAP) as coupling agents, at room temperature.
DMAP (30 mg), EDC (1.2 eq.) and the corresponding semi-fluorinated acid (1.2 eq.) were added to dichloromethane (10 mL). After stirring during 30 mn at room temperature, EDTPOH (1.0 eq) was added. The reaction mixture was stirred for 24 h at 50 °C. Then, the solvent was removed and the residue was purified by column chromatography (silica gel; eluent: dichloromethane) to yield the fluorinated monomers.
2.2. Electrodeposition experiments
The electropolymerization experiments were performed in anhydrous acetonitrile solutions containing 0.1 M of tetrabutylammonium hexafluorophosphate (Bu4NPF6) and 10 mM of monomer. The solutions were degassed before each experiment. These experiments were performed in a three-electrode cell composed of a graphite counter electrode, a saturated calomel reference electrode (SCE) and platinum disk or large gold plates as working electrode for cyclic voltammetry and chronoamperometry, respectively.2.3. Surface characterization
The arithmetic roughness (Ra) and rms roughness (Rq) were measured using a WYKO NT1100 optical profilometer on an area of 182 × 239 μm2. The surface morphology was investigated by scanning electron microscopy (SEM) using a JEOL 6700F. The surface wettability was characterized by static contact angle measurements (θw) with 2 μL water droplets and dynamic contact angle measurements (sliding angle, αw, and hysteresis, Hw) with 6 μL droplets using the tilted-drop method. Nucleation and growth mechanism study was performed using the software OriginPro 8.5. The models used were imported in the software and experimental curves were fitted using the non-linear curve fit option. Comparisons of various models were performed using the Akaike's Information Criteria.3. Results and discussion
3.1. Electrodeposition and electrochemical characterization
Using a platinum disk as working electrode, the monomer oxidation potentials, 0.88 V vs. SCE for EDTP-F4, EDTP-F6 and 0.87 V vs. SCE for EDTP-F8, were determined by cyclic voltammetry. Then, ten scans were performed until a potential slightly lower than the monomer oxidation to deposit a thin film of PEDTP-Fn on a Pt electrode tip. An example of a cyclic voltammogram is given in Fig. 1a for EDTP-F6. The three fluorinated monomers displayed similar cyclic voltammograms. After transferring the electrode into a monomer free solution, the curves obtained by cyclic voltammetry of the polymers showed similar shapes with a unique oxidation and reduction peak, as represented in Fig. 1b for PEDTP-F6. The oxidation/reduction potentials of these polymers were 0.72/0.71 V for PEDTP-F4, 0.75/0.72 V for PEDTP-F6 and 0.77/0.72 V for PEDTP-F8.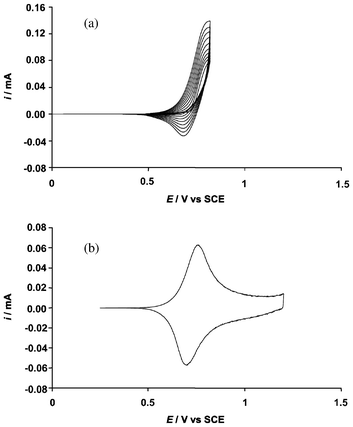 | ||
| Fig. 1 Cyclic voltammograms of (a) EDTP-F6 (10 scans) and (b) PEDTP-F6 (1 scan) in Bu4NPF6 (0.1 M) in acetonitrile. | ||
The polymer oxidation potentials were extremely close to that of the monomer, which means that the conjugation lengths of the polymers are very short. Hence, unlike poly(3,4-ethylenedioxypyrrole) derivatives,31 the presence of two sulfur atoms at the 3- and 4-positions suppresses the intramolecular interactions between adjacent units due to their size, as already reported in the literature.40,41 As a consequence, the planarity of the polymers is highly affected and the polymer length very reduced.
To determine the surface wettability and roughness, polymer films were electrodeposited on large gold plates by potentiostatic methods using various deposition charges (Qs) from 25 mC cm−2 to 1000 mC cm−2.
3.2. Surface wettability
The anti-liquid properties of the films were evaluated by measuring static and dynamic contact angles with water droplets. For Qs = 225 mC cm−2, these measurements with water revealed that PEDTP-F6 and PEDTP-F8 were superhydrophobic with static contact angles greater than 160° and very low sliding angles (α <5°) and hysteresis (H <10°), suggesting self-cleaning properties. PEDTP-F4 was also superhydrophobic but with a higher adhesion (θw = 155°, α = 27.5°, H = 26.2°). Two movies showing the difference between the adhesion of water droplet on PEDTP-F6 (low adhesion) and PEDTP-F4 (high adhesion) are given in the ESI.† These differences can be explained by the Wenzel and Cassie–Baxter theories.42,43 Indeed, in the Cassie–Baxter theory, the water droplet is suspended on the tops of the asperities and on air pockets. As a consequence, the contact angles are extremely high but the adhesion of the water on the surface is extremely low as well as the hysteresis and sliding angles. In the case of the Wenzel theory, the water droplet is in perfect contact with the surface and this time the surface roughness increases the contact angles but also the hysteresis.The electrodepositions were also performed at various deposition charges (Table 1). Unexpected results showed that, at low deposition charges, PEDTP-F6 showed greater hydrophobic properties than PEDTP-F8. It is therefore possible to reach better hydrophobic performances with shorter fluorinated chain lengths, by controlling the deposition process.
| Deposition charge Qs [mC cm−2] | PEDTP-F8 | PEDTP-F6 | PEDTP-F4 |
|---|---|---|---|
| 25 | 119 | 160 | 126 |
| 50 | 130 | 158 | 156 |
| 100 | 157 | 161 | 152 |
| 225 | 157 | 161 | 152 |
| 400 | 160 | 161 | 126 |
| 600 | 160 | 161 | 141 |
| 1000 | 160 | 160 | 142 |
3.3. Surface morphology and roughness
The exceptional water-repellency properties are probably due to the synergetic effect of the presence of surface roughness and morphology, as described by the Wenzel and Cassie–Baxter models,42,43 with the intrinsic hydrophobicity of polymers due to the introduction of fluorinated tails. SEM images of the polymers are gathered in Fig. 2 for Qs = 225 mC cm−2. The images showed deposits of smooth spherical particles of different sizes in the case of PEDTP-F4 and PEDTP-F6 while PEDTP-F8 displayed cauliflower-like microstructures. The range of particle sizes decreases with the increase in the fluorinated chain length (from about 1–5 μm for PEDTP-F4 to about 0.1–1 μm for PEDTP-F8). Only in the case of PEDTP-F4 was a significant increase of the particle size observed with the deposition charge (cf.Fig. 3: PEDTP-F4 for Qs = 1000 mC cm−2). Surprisingly, these structures were both hollow and compartmented as shown in Fig. 3. The largest structures for Qs = 1000 mC cm−2 collapsed due to their size and their hollow structure. Such hollow structures have already been obtained by oxidative chemical polymerization of aniline, pyrrole or carbazole, using templates.27–30,44,45 Tubular nanostructures were also obtained by electropolymerization of conducting polymers,46 but to the best of our knowledge, such hollow spheres have never been created through electropolymerization of conducting polymers. | ||
| Fig. 2 SEM images of (a and b) PEDTP-F4, (c and d) PEDTP-F6 and (e and f) PEDTP-F8; magnification × 250 and × 5,000; Qs = 225 mC cm−2. | ||
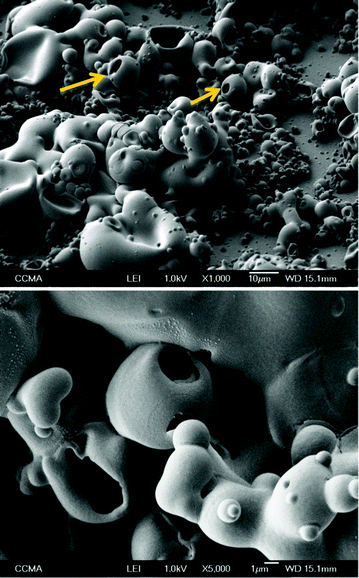 | ||
| Fig. 3 SEM images of electrodeposited PEDTP-F4; magnification × 1000 and × 5 000; Qs = 1000 mC cm−2. | ||
The roughness analyses allowed identification of two regimes of growth during potentiostatic depositions. After a nucleation period, arithmetic roughness (Ra) linearly increased to reach a maximum for PEDTP-F6 and PEDTP-F8 whereas PEDTP-F4 roughness continuously increased. These analyses point out the predominance of structure shape in wetting properties. Indeed, the maximum of roughness does not correspond to the best hydrophobic properties (high static contact angle and low hysteresis). Fig. 4 shows the optimal surface profile of PEDTP-F6 for superhydrophobicity. At low deposition charge, the surface roughness is too low to induce a Cassie–Baxter state (or “fakir state”)43 and at high deposition charge, the high surface roughness impedes liquid droplets rolling off the surface. Furthermore, the better wetting properties observed with PEDTP-F6 compared to PEDTP-F8 can be explained by the surface morphology. The microstructures formed at 25 and 50 mC cm−2 were more numerous, which probably provide a height/spacing ratio able to prevent the sagging of the water/air interface to the bottom of the microstructures.47 Thus the solid/liquid area is smaller for PEDTP-F6 than for PEDTP-F8, which explains the highest apparent contact angle, according to Cassie–Baxter predictions.43
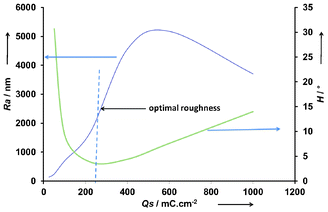 | ||
| Fig. 4 Arithmetic roughness and water contact angle hysteresis of PEDTP-F6. | ||
3.4. Effect of the fluorinated chain length by studying the nucleation and growth mechanisms
To determine the fluorinated chain length effect on the surface morphology, it is very important to have a good understanding of the process. Electrodeposition is a method known to produce various surface morphologies as a function of electrochemical parameters and many works in the literature showed these effects.48,49 Very recently, the group of Bendikov showed, by studying the electrodeposition of PEDOT in different solvents, that the surface structuring may increase with the insolubility of the PEDOT oligomers formed in the first instance.50In our case, because the solvent used is very polar (acetonitrile), the oligomer insolubility increases with polymer chain length and also with the fluorinated chain length. However, conjugation length of the polymer chains did not significantly increase with the fluorinated chain length, as demonstrated by cyclic voltammetry (the oxidation and reduction potentials were quite similar). Hence, the fluorinated chain length has probably an influence on the surface morphology because it changes the oligomer solubility produced in the first instances of the electrodeposition.
To confirm this assumption, the nucleation and growth mechanism (NGM) of PEDTP-F4 and PEDTP-F8 were studied by fitting current transient curves obtained during potentiostatic electrodeposition with theoretical models (Table 2).51 The nucleation can be instantaneous (IN) or progressive (PN) and the growth can be three dimensional (3D) or two dimensional (2D). The growth mechanism is controlled either by charge transfer (ct) or diffusion (dif). Thus, the current transient curves reflect either one of these behaviors or a combination of one to four contributions, each polymer having its own NGM. The literature describes the major route of film formation as follows: 1) initial oxidation of monomers which can be adsorbed on the electrode surface or coming from the solution and 2) the oxidized monomers diffuse towards the interface where the oligomerization takes place and, simultaneously, build an oligomeric high density region (OHDR). Once the supersaturation of the OHDR is attained, clusters are deposited on the electrode creating growing nuclei.51b
| Models | Mathematical expression |
|---|---|
| Induction | k·exp(−1·t) |
| IN2D | i·t·exp(−j·t2) |
| IN3Dct | a·(1 − exp(−b·t2)) |
| PN3Dct | c·(1 − exp(−d·t3)) |
| IN3Ddif |
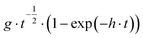
|
| PN3Ddif |
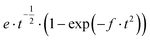
|
For PEDTP-F8, the model that fitted the best to experimental data was mainly composed of IN3Ddif and PN3Ddif contributions to the overall NGM whereas for PEDTP-F4, the model that fitted the best was mainly composed of IN3Dct and PN3Dct contributions. The deconvolution graphs are shown in Fig. 5. Hence, the growth of these two polymers is almost exclusively three-dimensional and the major difference is the kinetic control of the NGM. The diffusion control of PEDTP-F4 NGM is consistent with the high solubility of PEDTP-F4 oligomers. Indeed, many oligomers can be formed before the saturation of the OHDR. Thus, the monomer will have to cross this region before deposition. By contrast, the charge transfer control of PEDTP-F8 NGM is in good agreement with the low solubility of PEDTP-F8.
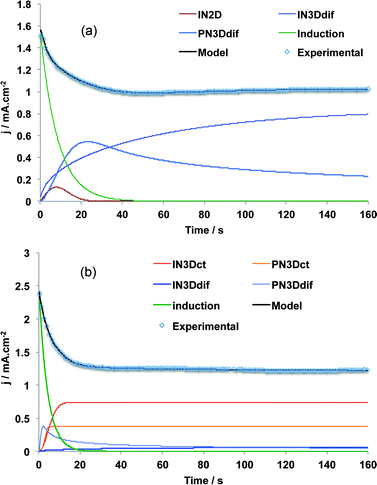 | ||
| Fig. 5 Deconvolution of the experimental current transient curves obtained for potentiostatic deposition of (a) PEDTP-F4 and (b) PEDTP-F8 on gold plates at Eopt. | ||
Profilometry images can confirm the validity of these models. Indeed, the predominance of progressive nucleation in the 40 first seconds of PEDTP-F4 deposition, which corresponds to a deposition charge of about 50 mC cm−2, explain the diversity of particle size observed, as shown on the 3D profilometry image and 2D profile in Fig. 6a–c. Here, the height of some particles could reach 6 μm. By contrast, the instantaneous nucleation is predominant after only 5 s in PEDTP-F8 NGM, which is consistent with the high homogeneity of particle size observed on the 3D profilometry image and 2D profile in Fig. 6b d. The weak 2D contribution during the 20 first seconds of PEDTP-F4 electrodeposition suggests that a thin layer of polymer covers the substrate although 3D particles are not present everywhere on the substrate.
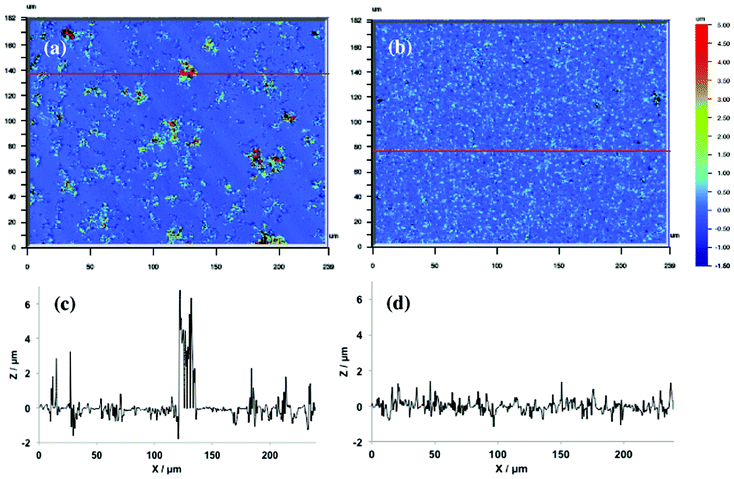 | ||
| Fig. 6 Profilometry images and 2D profiles of (a),(c) PEDTP-F4 and (b),(d) PEDTP-F8. | ||
This is confirmed visually by the disappearance of the mirror effect of the gold substrate after 20 s of deposition.
Hence, in the case of PEDTP-F8, very small and numerous polymer particles are formed during the first moments, which are instantaneously deposited on the substrate because of their high insolubility induced by long fluorinated tails (F-octyl). In the case of PEDTP-F4, few polymer particles are formed in the first instant, due to the higher solubility of oligomers, which explains the presence of some large spherical particles bearing smaller particles.
3.5. Effect of the polymer solubility on the hollow spheres formation
Polymer chains with short conjugation length were produced using these monomers, as observed by cyclic voltammetry, which is due to the presence of two sulfur atoms at the 3- and 4-positions of pyrrole. The solubility also increased along with the decrease of the fluorinated chain length. The increase in solubility changed the growth mechanism from instantaneous to progressive.All these experiments show that the conditions to produce hollow spheres seems to be: relative solubility of the electrodeposited polymers; three-dimensional growth, which is in part due to the presence of the fluorinated chain and the electrochemical parameters.
Indeed, the electrodeposition of conducting polymers often leads to the formation of compact spheres or assembly of spherical particles such as cauliflower-like structures. However, here, due to the presence of sulfur atoms and short fluorinated chains, the compacity between the polymer chains is probably highly reduced and the spheres are probably much more permeable to liquids and gas. Indeed, while the sulfur atoms highly reduce the polymer length and the interactions between them (confirmed by cyclic voltammetry), the interactions between F-butyl chains are extremely low in comparison with longer fluorinated chains.52 As a consequence, liquid or gas may penetrate inside the polymer and act as a soft template for the elaboration of hollow spheres.
Conclusions
Here, for the first time, hollow spheres were electrodeposited from fluorinated 3,4-ethylenedithiopyrrole. We have reported the synthesis and characterization of N-(2-hydroxyethyl)-3,4-ethylenedithiopyrrole (EDTP), as key synthon for the synthesis of fluorinated EDTP derivatives. The polymer films displayed superhydrophobic properties with a surface morphology consisting in assembly of spherical particles. Their mean size and the possibility of producing hollow structures increased with a decrease of the fluorinated chain length. The study of the growth by cyclic voltammetry and the use of theoretical nucleation and growth mechanism models showed that the determining parameters in the formation of hollow structures seem to be the presence of progressive three-dimensional growth associated with relative solubility of the polymers (short polymer chains and short fluorinated tails).References
- S.-W. Kim, M. Kim, W. Y. Lee and T. Hyeon, J. Am. Chem. Soc., 2002, 124, 7642 CrossRef CAS.
- H.-P. Liang, H.-M. Zhang, J.-S. Hu, Y.-G. Guo, L.-J. Wan and C.-L. Bai, Angew. Chem., Int. Ed., 2004, 43, 1540 CrossRef CAS.
- Y. Zhu, J. Shi, W. Shen, X. Dong, J. Feng, M. Ruan and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 5083 CrossRef CAS.
- Y. Vasquez, A. K. Sra and R. E. Schaak, J. Am. Chem. Soc., 2005, 127, 12504 CrossRef CAS.
- Y. Chang, J. J. Teo and H. C. Zeng, Langmuir, 2005, 21, 1074 CrossRef CAS.
- X. Sun and Y. Li, Angew. Chem., Int. Ed., 2004, 43, 3827 CrossRef CAS.
- Z. Dai, J. Zhang, J. Bao, X. Huang and X. Mo, J. Mater. Chem., 2007, 17, 1087 RSC.
- X.-L. Li, T.-J. Lou, X.-M. Sun and Y.-D. Li, Inorg. Chem., 2004, 43, 5442 CrossRef CAS.
- H.-J. Koo, Y. J. Kim, Y. H. Lee, W. I. Lee, K. Kim and N.-G. Park, Adv. Mater., 2008, 20, 195 CrossRef CAS.
- D. Wu, F. Zhu, J. Li, H. Dong, Q. Li, K. Jiang and D. Xu, J. Mater. Chem., 2012, 22, 11665 RSC.
- G. Zhang, L. Yu, H. B. Wu, H. E. Hoster and X. W. Lou, Adv. Mater., 2012, 24, 4609 CrossRef CAS.
- X. W. Lou, C. M. Li and L. A. Archer, Adv. Mater., 2009, 21, 2536 CrossRef CAS.
- M. A. Correa-Duarte, A. Kosiorek, W. Kandulski, M. Giersig and L. M. Liz-Marzan, Chem. Mater., 2005, 17, 3268 CrossRef CAS.
- D. G. Shchukin and R. A. Caruso, Chem. Mater., 2004, 16, 2287 CrossRef CAS.
- M. Sasidharan, N. Gunawardhana, H. N. Luitel, T. Yokoi, M. Inoue, S.-I. Yusa, T. Watari, M. Yoshio, T. Tatsumi and K. Nakashima, J. Colloid Interface Sci., 2012, 370, 51 CrossRef CAS.
- R. Dong, W. Liu and J. Hao, Acc. Chem. Res., 2012, 45, 504 CrossRef CAS.
- L. Zhao, F. Tao, Z. Quan, X. Zhou, Y. Yuan and J. Hu, Mater. Lett., 2012, 68, 28 CrossRef CAS.
- (a) S. Roy, A. Wanner, T. Beck, T. Studnitzky and G. Stephani, J. Mater. Sci., 2011, 46, 5519 CrossRef CAS; (b) I. Vida-Simiti, N. Jumate, E. Bruj, N. Sechel, G. Thalmaier, D. Nemes and M. Nicoara, Mater. Sci. Forum, 2011, 672, 141 CrossRef CAS.
- (a) F. He, P. Yang, D. Wang, C. Li, N. Niu, S. Gai and M. Zhang, Langmuir, 2011, 27, 5616 CrossRef CAS; (b) M.-P. Liu, C.-H. Li, H.-.B. Du and X.-Z. You, Chem. Commun., 2012, 48, 4950 RSC.
- (a) P.-J. Cai, Y.-J. Tang, Y.-T. Wang and Y.-J. Cao, Mater. Chem. Phys., 2010, 124, 10 CrossRef CAS; (b) J. Liu, Y. Yan, Z. Chen, Y. Gu and X. Liu, Chem. Lett., 2010, 39, 1194 CrossRef CAS.
- (a) S.-C. Luo, J. Jiang, S. S. Liour, S. Gao, J. Y. Ying and H.-H. Yu, Chem. Commun., 2009,(19), 2664 RSC; (b) H. Zhang, Y. Li, X. Wang, J. Li and F. Wang, Polymer, 2011, 52, 4246 CrossRef CAS; (c) M.-Y. Bai and Y. Xia, Macromol. Rapid Commun., 2010, 31, 1863 CrossRef CAS.
- (a) K. Liu, X. Yao and L. Jiang, Chem. Soc. Rev., 2010, 39, 3240 RSC; (b) M. Liu and L. Jiang, Adv. Funct. Mater., 2010, 20, 3753 CrossRef CAS.
- (a) K. Liu and L. Jiang, Nanoscale, 2011, 3, 825 RSC; (b) M. Liu, Y. Zheng, J. Zhai and L. Jiang, Acc. Chem. Res., 2010, 43, 368 CrossRef CAS.
- A. Lafuma and D. Quere, Nat. Mater., 2003, 2, 457 CrossRef CAS.
- Z. Guo, W. Liu and B.-L. Su, J. Colloid Interface Sci., 2011, 353, 335 CrossRef CAS.
- N. J. Shirtcliffe, G. McHale and M. I. Newton, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1203 CrossRef CAS.
- L. Tan, L. Cao, M. Yang, G. Wang and D. Sun, Polymer, 2011, 52, 4770–4776 CrossRef CAS.
- Y. Zhu, J. Li, M. Wan and L. Jiang, Macromol. Rapid Commun., 2008, 29, 239 CrossRef CAS.
- Y. Zhu, D. Hu, M. X. Wan, L. Jiang and Y. Wei, Adv. Mater., 2007, 19, 2092 CrossRef CAS.
- W. Zhong, Y. Li, Y. Wang, X. Chen, Y. Wang and W. Yang, J. Colloid Interface Sci., 2012, 365, 28 CrossRef CAS.
- T. Darmanin and F. Guittard, J. Am. Chem. Soc., 2009, 131, 7928 CrossRef CAS.
- H. Bellanger, T. Darmanin and F. Guittard, Langmuir, 2012, 28, 186 CrossRef CAS.
- (a) T. Darmanin and F. Guittard, J. Am. Chem. Soc., 2011, 133, 15627 CrossRef CAS; (b) M. Wolfs, T. Darmanin and F. Guittard, Macromolecules, 2011, 44, 9286 CrossRef CAS.
- (a) T. Darmanin, E. Taffin de Givenchy and F. Guittard, Macromolecules, 2010, 43, 9365 CrossRef CAS; (b) M. Wolfs, T. Darmanin and F. Guittard, Soft Matter, 2012, 8, 9110 RSC.
- (a) S.-C. Luo, J. Sekine, B. Zhu, H. Zhao, A. Nakao and H.-H. Yu, ACS Nano, 2012, 6, 3018 CrossRef CAS; (b) S.-C. Luo, S. S. Liour and H.-H. Yu, Chem. Commun., 2010, 46, 4731 RSC.
- (a) L. Xu, W. Chen, A. Mulchandani and Y. Yan, Angew. Chem., Int. Ed., 2005, 44, 6009 CrossRef CAS; (b) H. Yan, K. Kurogi, H. Mayama and K. Tsujii, Angew. Chem., Int. Ed., 2005, 44, 3453 CrossRef CAS.
- J. O. Jeppesen, K. Takimiya, F. Jensen, T. Brimert, K. Nielsen, N. Thorup and J. Becher, J. Org. Chem., 2000, 65, 5794 CrossRef CAS.
- H. Li, C. Lambert and R. Stahl, Macromolecules, 2006, 39, 2049 CrossRef CAS.
- J. A. Schiffner, T. H. Woeste and M. Oestreich, Eur. J. Org. Chem., 2010, 174 CrossRef CAS.
- (a) T. A. Skotheim and J. R. Reynoldsin Handbook of Conducting Polymers, Third Edition; Conjugated Polymers: Theory, Synthesis, Properties and Characterization, CRC Press Taylor & Francis Group, Boca Raton, USA 2007 Search PubMed; (b) H. J. Spencer, P. J. Skabara, M. Giles, I. McCulloch, S. J. Coles and M. B. Hursthouse, J. Mater. Chem., 2005, 15, 4783 RSC.
- (a) M. Turbiez, P. Frere, M. Allain, N. Gallego-Planas and J. Roncali, Macromolecules, 2005, 38, 6806 CrossRef CAS; (b) J.-M. Raimundo, P. Blanchard, P. Frere, N. Mercier, I. Ledoux-Rak, R. Hierle and J. Roncali, Tetrahedron Lett., 2001, 42, 1507 CrossRef CAS.
- R. N. Wenzel, Ind. Eng. Chem., 1936, 28, 988 CrossRef CAS.
- (a) A. B. D. Cassie and S. Baxter, Trans. Faraday Soc., 1944, 40, 546 RSC; (b) S. Baxter and A. B. D. Cassie, J. Text. Inst., Trans., 1945, 36, T67 CrossRef CAS.
- (a) M. Yang, X. X. Yao, G. Wang and H. Ding, Colloids Surf., A, 2008, 324, 113 CrossRef CAS; (b) Y. S. Zhang, W.-H. Xu, W.-T. Yao and S.-H. Yu, J. Phys. Chem. C, 2009, 113, 8588 CrossRef CAS; (c) J. Stejskal, I. Sapurina, M. Trchova and E. N. Konyushenko, Macromolecules, 2008, 41, 3530 CrossRef CAS; (d) L. Zhang and M. Wan, Adv. Funct. Mater., 2003, 13, 815 CrossRef CAS.
- (a) S.-H. Choi, A. I. Gopalan, J.-H. Ryu and K.-P. Lee, Mater. Chem. Phys., 2010, 120, 18–22 CrossRef CAS; (b) M. J. Antony and M. Jayakannan, J. Phys. Chem. B, 2010, 114, 1314 CrossRef CAS.
- B. Gupta and R. Prakash, Synth. Met., 2010, 160, 523 CrossRef CAS.
- (a) C. W. Extrand, Langmuir, 2002, 18, 7991 CrossRef CAS; (b) C. W. Extrand, Langmuir, 2006, 22, 1711 CrossRef CAS; (c) C. W. Extrand, Langmuir, 2004, 20, 5013 CrossRef CAS.
- (a) R. J. Mammone and M. Binder, J. Electrochem. Soc., 1990, 137, 2135 CrossRef CAS; (b) G. A. Wood and J. O. Iroh, Polym. Eng. Sci., 1996, 36, 2389 CrossRef CAS; (c) T. Silk, Q. Hong, J. Tamm and R. G. Compton, Synth. Met., 1998, 93, 59 CrossRef CAS.
- (a) K. M. Cheung, D. Bloor and G. C. Stevens, Polymer, 1988, 29, 1709 CrossRef CAS; (b) W. Su and J. O. Iroh, Synth. Met., 1998, 95, 159 CrossRef CAS; (c) T. F. Otero and E. DeLaretta, Synth. Met., 1988, 26, 79 CrossRef CAS.
- E. Poverenov, M. Li, A. Bitler and M. Bendikov, Chem. Mater., 2010, 22, 4019 CrossRef CAS.
- (a) J. P. Soto, F. R. Diaz, M. A. del Valle, J. H. Velez and G. A. East, Appl. Surf. Sci., 2008, 254, 3489 CrossRef CAS; (b) M. A. del Valle, P. Cury and R. Schrebler, Electrochim. Acta, 2002, 48, 397 CrossRef CAS; (c) R. Schrebler, P. Grez, P. Cury, C. Veas, M. Merino, H. Gomez, R. Cordova and M. A. del Valle, J. Electroanal. Chem., 1997, 430, 77 CrossRef CAS; (d) H. Randriamahazaka, V. Noel and C. Chevrot, J. Electroanal. Chem., 1999, 472, 103 CrossRef; (e) H. Randriamahazaka, G. Sini and F. Tran Van, J. Phys. Chem. C, 2007, 111, 4553 CrossRef CAS.
- K. Honda, M. Morita, H. Otsuka and A. Takahara, Macromolecules, 2005, 38, 5699 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Mass spectra and movies. See DOI: 10.1039/c2ra21665e |
| This journal is © The Royal Society of Chemistry 2012 |