DNA glycoclusters and DNA-based carbohydrate microarrays: From design to applications†
François
Morvan
*a,
Sébastien
Vidal
c,
Eliane
Souteyrand
b,
Yann
Chevolot
*b and
Jean-Jacques
Vasseur
*a
aInstitut des Biomolécules Max Mousseron (IBMM), UMR 5247 CNRS - Université Montpellier 1 - Université Montpellier 2, place Eugène Bataillon, CC1704, 34095 Montpellier cedex 5, France. E-mail: vasseur@univ-montp2.fr; Fax: +33 467 042 029; Tel: +33 467 143 320 morvan@univ-montp2.fr; Tel: +33 467 144 961
bInstitut des Nanotechnologies de Lyon (INL), UMR 5270 CNRS Ecole Centrale de Lyon, 36 Avenue Guy de Collongue, 69134 Ecully cedex, France. E-mail: yann.chevolot@ec-lyon.fr; Fax: +33 478 433 593; Tel: +33 472 186 240
cInstitut de Chimie et Biochimie Moléculaires et Supramoléculaires (ICBMS), Laboratoire de Chimie Organique 2 – Glycochimie, UMR 5246, CNRS, Université Claude Bernard Lyon 1, 43 Boulevard du 11 Novembre 1918, 69622 Villeurbanne, France
First published on 17th September 2012
Abstract
Our goal was to design carbohydrate mimetics capable of preventing cell adhesion of pathogens by targeting lectins. The original synthesis of these mimetics combines the automated chemistry of DNA and “click” chemistry. From simple building blocks (i.e. phosphoramidites, solid supports and carbohydrates such as alkyne or azide derivatives), a “Lego” approach provides complex and varied decoys exhibiting various topologies and number of carbohydrate residues. The resulting glycomimetics tagged with a specific DNA sequence are efficiently immobilized on a DNA microarray by double strand formation. This glycoarray allows the study of the interactions between carbohydrate mimics and lectins using a minute amount of material. This micro system using DNA arrays is much more effective than conventional carbohydrate microarrays. The studies allowed the identification of the important structural parameters for a customized construction of high-affinity carbohydrate mimics.
 François Morvan | François Morvan received his PhD in organic chemistry (1988) at the University of Montpellier 2 (France) under the supervision of Dr Rayner and Prof. Imbach on the synthesis of alpha-DNA. In 1989, he was hired at the Inserm working on modified oligonucleotides for antisense applications. He was senior visiting scientist at ISIS Pharmaceuticals (USA) from 1992 to 1993. He then returned to the University of Montpellier 2 and is currently Research Director at the Inserm. His research focuses on modified oligonucleotides for diagnostic and therapeutic applications, and design of glycoclusters for anti-adhesive strategies against bacterial infections. |
 Sébastien Vidal | Sébastien Vidal received his PhD in Organic Chemistry (2000) at University of Montpellier (France) under the supervision of Professor Montero and synthesized mannose 6-phosphate analogues. He then moved to the group of Professor Stoddart at UCLA for the synthesis and characterization of glycodendrimers. In 2003, he joined the National Renewable Energy Laboratory with Prof. Bozell to investigate the combination of organometallic and carbohydrate chemistries. In 2004, he obtained a CNRS position at University of Lyon. The main topics covered in his research are the design of glycoclusters for anti-adhesive strategies against bacterial infections but also enzyme inhibitors. |
 E. Souteyrand | E. Souteyrand obtained her PhD degree (1979) and “Docteur d'Etat” in Materials Sciences (1985) from Paris VI University. She joined CNRS (1981), specializing in electrochemistry and semiconducting/dielectric/liquid interfaces; she investigated the field of chemical and biosensors, and in particular DNA chips. In 2000, she received the Award for Innovative Company Creation and founded Rosatech S.A., a company devoted to “biochips for diagnostics”. Currently, she leads the “Chemistry and NanoBiotechnology“ group at the Institute of Nanotechnology of Lyon. She is the co-author of more than 100 international publications, and holds 9 international patents. |
 Yann Chevolot | Yann Chevolot received his PhD degree in material science from the Ecole Polytechnique Fédérale de Lausanne at Lausanne (Switzerland) in 1999. Between 1999 and 2001, he worked as assistant of Prof. Mathieu at EPFL on bacterial adhesion to PVC endotracheal tubes and was in charge of ToF-SIMS analysis. From 2001 to 2004, he worked in the research department of Goemar SA, Saint Malo (France). In 2004, he joined the CNRS as a senior scientist. He focuses on microfabrication, surface chemistry and characterisation, and biochips, in particular glycoarrays. |
 Jean-Jacques Vasseur | Jean-Jacques Vasseur completed his PhD studies in organic chemistry (1988) working on DNA abasic sites with Prof. Imbach and Dr Rayner at the University of Montpellier (France). From 1990 to 1992, he was a visiting scientist at Isis Pharmaceuticals (Carlsbad, CA, USA) developing oligonucleotides without phosphate. In 1993, he went back to Montpellier with a CNRS position. He is currently Research Director at the CNRS, leader of the Department of Nucleic Acids in the Institute of Biomolecules Max Mousseron. His research focuses on nucleic acid chemistry and recognition for diagnostic and therapeutic applications. |
Introduction
Carbohydrates associated with biomolecules such as proteins (glycoproteins) and lipids (glycolipids) are part of the high molecular density layer called the glycocalix covering the surface of epithelial cells in eukaryotes. The interactions of these carbohydrates with proteins play a critical role in many biological processes such as cellular communication, inflammation due to interplay with antibodies, and infections by pathogens. The binding of bacteria and viruses to natural glycoconjugates at the surface of the host cell is considered a key step for infection. Proteins involved in these recognition processes are called lectins but could be also toxins. The study of these interactions is fundamental for understanding the mechanism of adhesion of pathogens, and to possibly develop inhibitors as new therapeutic agents, but also to detect the pathogen.The chemical synthesis of carbohydrates and consequently the analysis of their interactions with proteins such as lectins are far from simple due to the high diversity and complexity of oligosaccharides when compared to other biopolymers such as nucleic acids and proteins.1,2 In nucleic acids, from the four deoxynucleotides bearing two possible elongation sites (5′- and 3′-O), 46 = 4096 different linear hexamers can be formed, where the units are connected through phosphodiester bonds. For proteins, due to the larger number of natural amino acids (20), bearing here again two elongation sites (COOH and NH2), there are 206 = 64![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 possibilities of linear hexapeptides where the units are linked through amide bonds. The number of structural combinations for carbohydrates is much higher; first because of the diversity of the saccharidic units: hexoses, pentoses with furanose or pyranose forms, N-acylated saccharides, sialic acids, deoxy but also sulfated, methylated and phosphorylated saccharides, and second because of a greater number of possible elongation sites on a monosaccharide. Thus, considering a saccharide-like galactose in a pyranose form, there are 5 possible elongation sites (1-, 2-, 3-, 4-, and 6-OH), so linear as well as branched oligomers can be formed. To this unique feature, a further complexity is added by the creation of a stereogenic center (α or β) during the formation of the glycosidic bond between saccharidic residues. Thus, starting from the most 10 common mammalian monosaccharides, 192780943360 hexasaccharides could be formed.3,4 Besides, compared to the synthesis of peptides and oligonucleotides which are now well-established, robust and automated, the chemical synthesis of oligosaccharides is still challenging, difficult and time-consuming.5–7 For this reason access to pure and complex oligosaccharides is limited, but analytical methods to study their interactions with lectins require relative large quantities of material.
000000 possibilities of linear hexapeptides where the units are linked through amide bonds. The number of structural combinations for carbohydrates is much higher; first because of the diversity of the saccharidic units: hexoses, pentoses with furanose or pyranose forms, N-acylated saccharides, sialic acids, deoxy but also sulfated, methylated and phosphorylated saccharides, and second because of a greater number of possible elongation sites on a monosaccharide. Thus, considering a saccharide-like galactose in a pyranose form, there are 5 possible elongation sites (1-, 2-, 3-, 4-, and 6-OH), so linear as well as branched oligomers can be formed. To this unique feature, a further complexity is added by the creation of a stereogenic center (α or β) during the formation of the glycosidic bond between saccharidic residues. Thus, starting from the most 10 common mammalian monosaccharides, 192780943360 hexasaccharides could be formed.3,4 Besides, compared to the synthesis of peptides and oligonucleotides which are now well-established, robust and automated, the chemical synthesis of oligosaccharides is still challenging, difficult and time-consuming.5–7 For this reason access to pure and complex oligosaccharides is limited, but analytical methods to study their interactions with lectins require relative large quantities of material.
With such diversity, one can figure out that the recognition of oligosaccharides with lectins is also complex. Actually, it involves a network of weak energy interactions participating in a high-density coding system.8 As a well-known example, the interaction of the T-antigen (Galβ(1->3)GalNAc) with peanut agglutinin (PNA) involves direct hydrogen bonds mostly with the non reducing galactose unit of the disaccharide, and H-bonds through several water bridging molecules (23 identified). Beside these H-bonds there are a plethora of hydrophobic linkages — mainly van der Waals interactions (60 identified) and a strong π–π stacking between the hydrophobic α-face of the non-reducing galactose and a tyrosine residue of the lectin.9 These multiple interactions provide the specificity of recognition in which the non-reducing unit is essential. However, due to the weakness of these interactions, monosaccharides usually bind to lectins with low affinity. To compensate this lack of strength, Nature uses multivalency. A lectin usually has several binding sites interacting simultaneously with several oligosaccharides on the cell surface. For example, the cholera toxin contains five B-subunits with five individual binding sites each interacting with a GM1 oligosaccharide on the intestinal cell surface.10 As another example, the soluble lectin PA-IL of the bacterium Pseudomonas aeruginosa consists of four sub-units associated as a tetramer able to interact specifically with four D-galactose units.11,12 Last, in the hemagglutinin of influenza viruses, three subunits associate to form trimers and interact with three sialic acids of sialylated lactosides at the cell surface of bronchial epithelial cells. This multivalent presentation compensates for the weakness of an individual binding event and allows the binding to become sufficiently strong for an efficient attachment of the virus to the host cell.13
Emulating the natural multivalency related to these interactions, chemists have surmised that the attachment to the cells could be blocked by multivalent synthetic glycoconjugates constituted of simple saccharidic units attached to various chemical frames and scaffolds.14–17 In addition, polyvalent interactions can be stronger than corresponding monovalent contributions provided that the orientation and spacing of the saccharide units on their scaffold match the binding sites of the lectin. If this proper presentation is met, a so-called cluster effect is observed resulting in an enhancement in the affinity of the ligand, higher than what would be expected with an increase in the concentration of saccharide only.18,19 However, while most glycomimetics reported in the literature are concerned with multivalency, few of them care about geometry constraints. Indeed, it is difficult to predict what could be the optimal glycomimetic scaffold for a given lectin. Therefore, it is often necessary to test several structures to find the best. In addition, most of these structures involve conventional chemistries. Due to the amount of compound required to study the interaction between glycomimetics and lectins using standard analytical techniques such as inhibition of agglutination of red blood cells (HIA), Enzyme Linked Lectin Assays (ELLA), Isothermal Titration Calorimetry (ITC), Surface Plasmon Resonance (SPR), chemical syntheses could be time- and money-consuming to obtain and study a diversity of glycomimetics. In comparison to these conventional analytical methods, carbohydrate microarrays allow the qualitative and quantitative study of several binding events at the same time on a single microarray with tiny amounts of materials (usually picomoles of material).20–22
We would like to show in this review that DNA chemistry used for the synthesis of glycomimetics combined with the use of microarrays to analyze the interactions between these glycoconjugates and lectins is a method of choice to quickly obtain a wide variety of architectures in sufficient quantities for the study of interactions on miniaturized DNA glycoarrays. These DNA glycomimetics consist of a scaffold obtained using standard DNA chemistry conjugated with saccharidic units. We will show that DNA glycoarrays relying on DNA Directed Immobilization (DDI) had several advantages over conventional immobilization techniques.
I. DNA chemistry is a versatile method to synthesize glycomimetics
I.1. Sequential approach and post-elongation conjugation
The power of DNA chemistry derives from an ideal combination of the efficacy of the phosphoramidite method first reported by Beaucage, Matteucci and Caruthers in 198123,24 combined with the efficiency of solid-phase synthesis25 associated to automation with the help of DNA synthesizers. A standard DNA synthesis cycle involves 4 steps (Fig. 1). In a first step, a nucleoside unit anchored through its 3′-OH to a solid support and protected at the 5′ position with a DMTr protecting group is detritylated in acidic medium to release a 5′-OH group which is then coupled in a second step to a protected nucleotide 3′-phosphoramidite activated by an acidic activator such as tetrazole. The nearly quantitative yield of the coupling step is obtained using a 10–20-fold excess of the phosphoramidite reagent with respect to the supported nucleotide. This reaction gives rise to a supported dinucleotide with a phosphite triester internucleoside linkage which is oxidized with iodine/water in a third step into a more stable phosphotriester linkage. In a fourth step, the unreacted 5′-OH moieties are capped with acetic anhydride. The cycle is then repeated as many times as necessary to obtain a protected oligonucleotide of desired sequence and length. Average yield per cycle is generally estimated to be superior to 99%. The release of the oligonucleotide from the solid support and its deprotection are performed with aqueous ammonia if standard protecting groups such as acyl and cyanoethyl are used for the nucleobase and phosphate protections, respectively.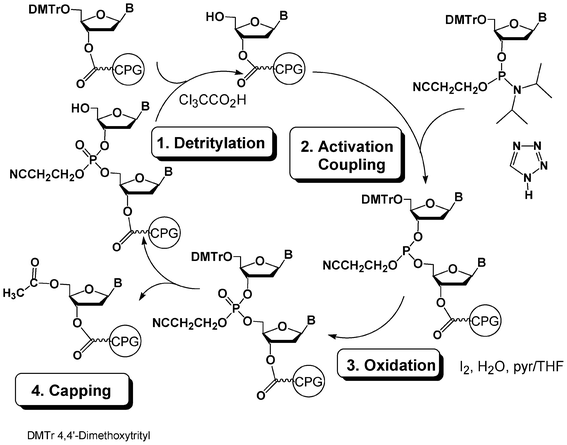 | ||
| Fig. 1 DNA synthesis cycle using phosphoramidite chemistry. | ||
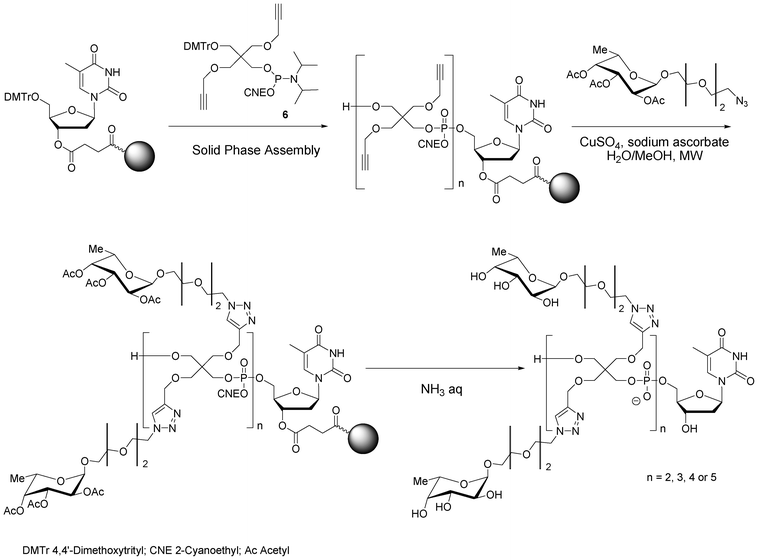
A similar synthesis cycle could be exploited for the synthesis of glycomimetics by replacing the solid-supported and phosphoramidite DMTr nucleoside building blocks with solid-supported and phosphoramidite saccharide units (Fig. 2). Solid-supported synthesis of DNA glycoconjugates using such an approach has been reviewed by Lönnberg in 200926,27 and, thus will be briefly described here. In this process, the saccharides are introduced sequentially in a phosphodiester backbone during the elongation on a solid support with a DNA synthesizer.
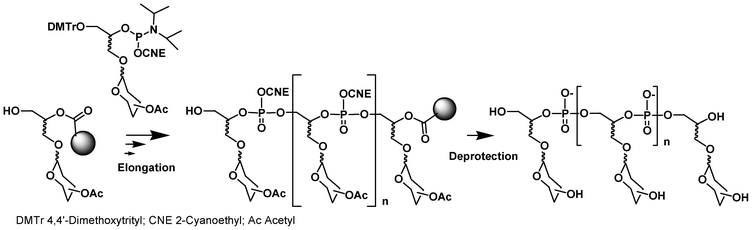 | ||
| Fig. 2 Synthesis of glycoclusters by phosphoramidite chemistry. | ||
Kobayashi et al.28,29 reported kind of a hybrid chemical strategy using nucleotide building blocks with nucleobases functionalized with a saccharide moiety (Fig. 3). This approach involves DNA chemistry but also uses the structure of DNA to organize the sugars along the DNA double helix.
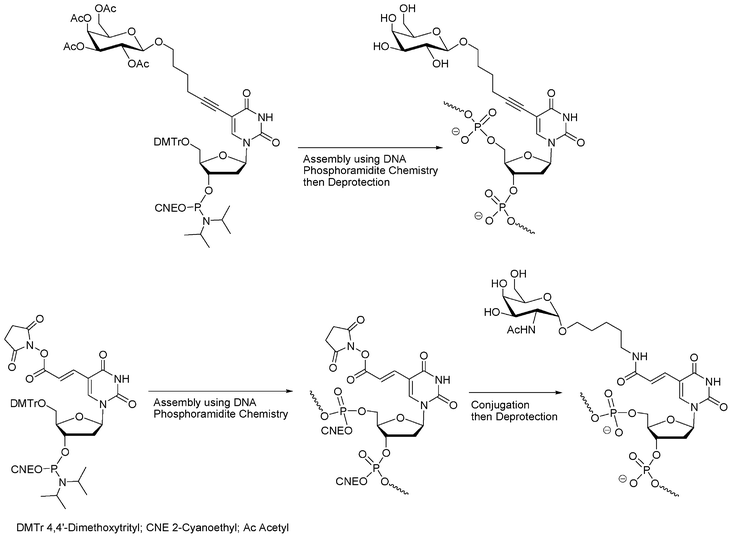 | ||
| Fig. 3 Synthesis of oligonucleotide glycoconjugates using modified nucleoside phosphoramidites with saccharide introduction performed before28,29 or after DNA elongation.30 | ||
While moving away from DNA chemistry for the synthesis of glycoconjugates, this unique hybridization property of nucleic acids was more recently developed to produce a collection of architectures from PNA–oligosaccharide conjugates with complementary DNA templates to mimic the topology of complex carbohydrates31,32 and allow the spatial screening of carbohydrate–lectin interactions.33,34 Finally, this use of the DNA double helix as a scaffold for a spatially definite presentation of carbohydrate antigens as potential ligands for lectins was reported by Seeberger.30 In this approach, the carbohydrates were conjugated to the solid-supported oligonucleotide in a post-conjugation approach (Fig. 3).
Instead of a nucleoside–saccharide conjugate, the carbohydrate could itself bear the phosphoramidite function. If all the hydroxyl functions of the saccharide building block are protected (except the one bearing the phosphoramidite function), these building blocks could be incorporated onto the 5′-end of a supported oligonucleotide, either directly35,36 or through various linkers assembled on solid supports as non-nucleosidic phosphoramidites acting as spacer and branching units, creating a diversity of possible glycosidic glycodendrimers from simple building blocks (Fig. 4).37,38
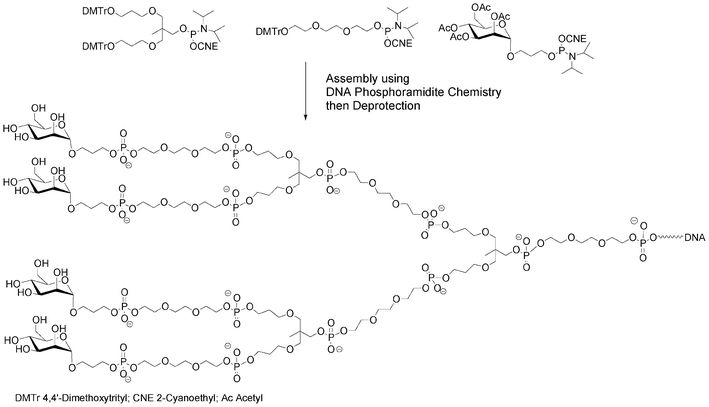 | ||
| Fig. 4 Synthesis of a mannosylated glycodendrimer using solid-phase DNA-like synthesis.37 | ||
A strategy combining nucleosidic, non-nucleosidic and non-saccharidic phosphoramidite units and saccharidic units was employed by Lönnberg39 for the synthesis of oligonucleotide glycoconjugates carrying three different saccharides. A phosphoramidite unit bearing three hydroxyl functions protected by the orthogonal levulinyl (Lev), tert-butyldiphenylsilyl (TBDPS) and 4,4′-dimethoxytrityl groups (Fig. 5) was coupled to an oligonucleotide at the end of the elongation cycle on a solid support and treated as a normal phosphoramidite building block. Thus, after oxidation, the DMTr group was cleaved, the hydroxymethyl elongated with one T-phosphoramidite before the introduction of a first saccharidic 6-O-phosphoramidite unit (Fig. 5,  ). The Lev protection was removed and a second saccharidic 6-O-phosphoramidite was coupled (Fig. 5,
). The Lev protection was removed and a second saccharidic 6-O-phosphoramidite was coupled (Fig. 5,  ). Finally the TBDPS was removed and a third 6-O-glycosyl phosphoramidite was reacted (Fig. 5,
). Finally the TBDPS was removed and a third 6-O-glycosyl phosphoramidite was reacted (Fig. 5,  ).
).
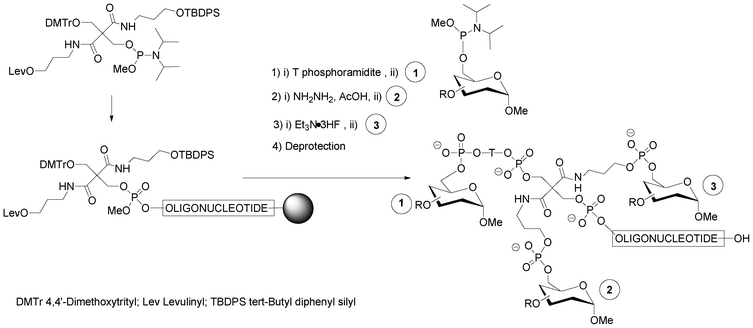 | ||
| Fig. 5 Solid-phase phosphoramidite assembly of oligonucleotide glycoconjugates displaying three different saccharidic units.39 | ||
If one of the hydroxyl functions of the saccharide phosphoramidite building block is DMT protected, then the synthon can be treated like a normal nucleotide monomer during the solid phase DNA assembly. This enables the sequential addition of a defined number of saccharidic phosphodiesters residues tethered40 or not41 to an oligonucleotide sequence. Carbohydrates are then linked inside or at the end of the construction through phosphodiester bonds.36 Noticeably, Montesarchio et al.41 were able at the end of the phosphoramidite elongation to cyclize a phosphate-linked oligosaccharide containing glucose units using phosphotriester methodology (Fig. 6). For this purpose, the free OH saccharidic end was coupled to the saccharide unit linked to the solid-support through a 2-chlorophenylphosphodiester linkage under activation with 1-mesitylensulfonyl-3-nitro-1,2,4-triazole and the cyclic oligosaccharide cleaved using oximate ions.42
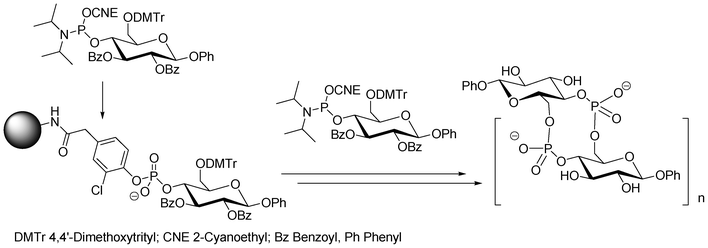 | ||
| Fig. 6 Solid-phase DNA-like synthesis of cyclic phosphate-linked oligosaccharides using bifunctional saccharidic units as building blocks.41 | ||
More complex carbohydrates could also be used as building blocks, i.e. phosphoramidite and solid supported units. Thus, O-DMTr solid-supported sucrose or O-DMTr sucrose phosphoramidite units give rise to 3′-terminal or 5′-terminal oligonucleotide glycoconjugates, respectively. These two units used in association gave oligonucleotides decorated with sucrose on both 5′- and 3′-ends.43 2′-O-Methyl oligoribonucleotides decorated with ribostamycin, an aminoglycoside having ribonuclease activity, were prepared in order to obtain specific artificial nucleases. These glycoconjugates were obtained by introduction inside the sequence of non-nucleosidic ribofuranose phosphoramidite units bearing protected oxyalkyl side arms (Fig. 7). The deprotection of these side chains was followed by the coupling of a ribostamycin phosphoramidite.44
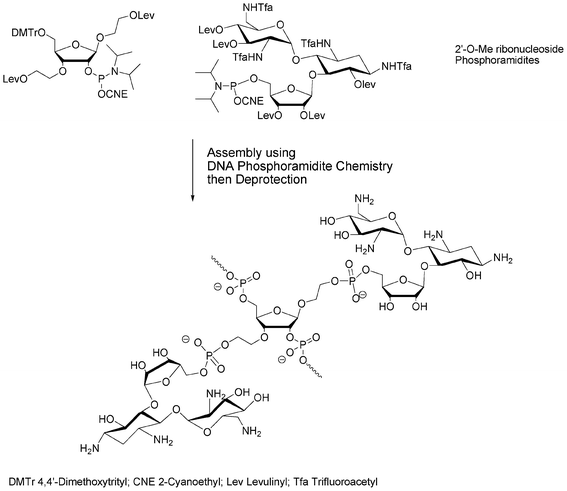 | ||
| Fig. 7 Solid phase phosphoramidite assembly of 2′-O-Me RNA conjugates of ribostamycin.44 | ||
The main advantage of this strategy is to use saccharide building blocks as nucleoside building blocks and to consider both saccharide and nucleoside elongation processes as similar. The main disadvantage, from our point of view, results from the diversity of saccharides and their multiple possible sites of derivatisation requiring well-thought-out protection–deprotection strategies that could be different from one to another saccharide unit. This could be tedious and expensive considering that large excesses of these extremely dedicated building blocks are needed for incorporation into the pseudo-oligonucleotidic chain.
This is the reason why we have favoured a post-elongation conjugation approach instead of introduction during the elongation process. Solid-supported DNA synthesis was used to construct a molecular scaffold bearing functions that could react at the end of the elongation process with appropriate functions carried by the saccharides, either on a solid support before release of the glycoconjugate from the support or in solution after cleavage of the scaffold from the solid support (Fig. 8). According to this strategy it is possible for the same scaffold to synthesize various glycomimetics by simply choosing the appropriate saccharide unit. This is a clear advantage of the post elongation conjugation compared to the sequential approach described before since usually the same carbohydrate was required to be added several times. The requisite for this post-elongation conjugation approach is a need for an extremely efficient and chemoselective reaction to conjugate and polyconjugate saccharides to these multifunctional scaffolds.
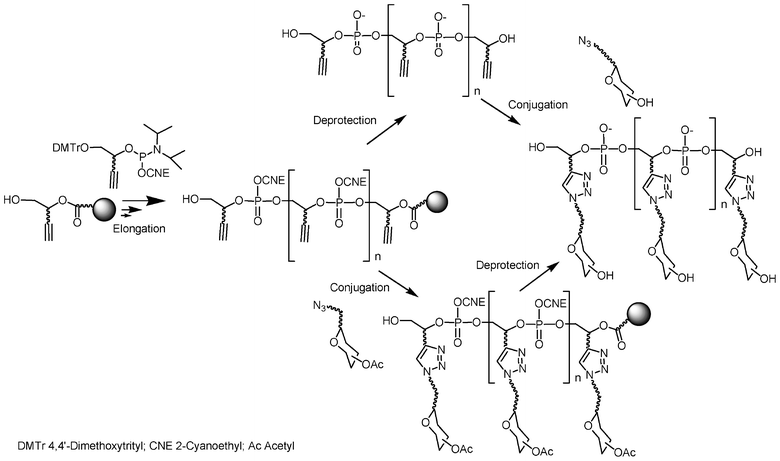 | ||
| Fig. 8 Synthesis of glycoclusters by combination of phosphoramidite chemistry and “click” chemistry. | ||
I.2. click chemistry as a tool for multiconjugation
The chemical reaction chosen for the conjugation must generate the desired glycoconjugates in high yields, with chemo- and regio-selectivity and must be consistent with aqueous media especially if the conjugation is performed after deprotection of the DNA-like phosphodiester scaffold with unprotected saccharide units. The concept of the “click reaction”, postulated by Sharpless in 200145 is related to such reactions. Since this report, there has been huge development of this principle in numerous applications and bioconjugation reaction. From the pioneering works of Huisgen on 1,3-dipolar cycloaddition in 1963,46 the copper(I)-catalyzed azide alkyne 1,3-dipolar cycloaddition (CuAAC or click-H) yielding only 1,4-disubstituted 1,2,3-triazoles47,48 is definitely the most extensively exploited click reaction and has become so popular that it is often equated to click reaction.49 This is also true for glycoconjugates.50–53 CuAAC is not the only click reaction utilized for saccharide conjugation. The reaction between an aldehyde and an aminooxy to give rise to an oxime, known as click-oxime (click-O), has been used several times in the field.54–59It is generally argued that one advantage of the CuAAC reaction is the tolerance of the azide and alkyne functions to a broad diversity of organic functions and reaction conditions. Thus, the synthesis of DNA-glycoconjugates through CuAAC can be theoretically achieved either from alkynyl-functionalized saccharides with a DNA-like scaffold bearing azido functions or from azido-functionnalized saccharides with a DNA-like scaffold bearing alkynyl functions. However, it is know that azido alkanes can react with phosphoramidites60 to lead to phosphorodiamidites and with phosphite triesters61 to give rise to phosphoramidates in the so-called Staudinger-phosphite reaction. As phosphoramidites and phosphite triesters are functional groups involved in DNA chemistry following the phosphoramidite approach, the synthesis of a building block bearing both azide and phosphoramidite functionalities is not conceivable since it decomposed in solution.62 However, it is feasible to introduce an azido function into a DNA-like scaffold using a pro-azide function such as bromo or iodo functions.63–66 Another consideration is that it has been recently demonstrated that an activated phosphoramidite can be coupled to an alcohol in presence of an azido function because the coupling is faster than the Staudinger reaction slowed down in acidic medium.67 Then, the expected phosphotriester compound is obtained provided that the phosphite triester is immediately oxidized into a more stable phosphate triester. The consequence is that solid supports functionalized with an azido function can be also relevant in our approach (vide supra). If there are some restrictions due to the peculiar reactivity of phosphoramidites and phosphites with azido compounds, this is not case for hydrogenophosphonate mono- and diesters. Indeed Lönnberg had reported the use of nucleoside H-phosphonates bearing azido functions following the H-phosphonate methodology (vide infra).68,69
I.3. A tool box for lego chemistry: non-nucleosidic phosphoramidite building blocks and solid supports
The alkyne, azide or pro-azide functions could be either introduced through nucleosidic or non-nucleosidic building blocks. Lönnberg et al. used the nucleosidic approach following the H-phosphonate methodology to bring azido functions to the 4′- or 2′-positions of an oligonucleotide (Fig. 9).68,69 Similarly, the same group introduced alkyne functions into an oligonucleotide using an appropriate 4′-C-alkyne modified nucleoside phosphoramidite synthon (Fig. 9).70 The aim was to enhance cell penetration and cell recognition by decoration of the oligonucleotides with simple (mannose) or more complex saccharides (paromamine and neomycin) through interactions with membrane bound lectins. The phosphoramidite building block 4′-C-[N,N-di(4-pentyn-1-yl)aminomethyl]thymidine containing two alkyne functions was employed to prepare oligonucleotides containing up to four mannose ligands by “click” on solid support. Introduction of alkyne functions at the 2′-positions of oligoribonucleotides in order to conjugate them with various azido ligands including saccharides was also reported by Manoharan et al. to study the effect of conjugation in 2′-O-position for siRNA therapeutic screening (Fig. 9).71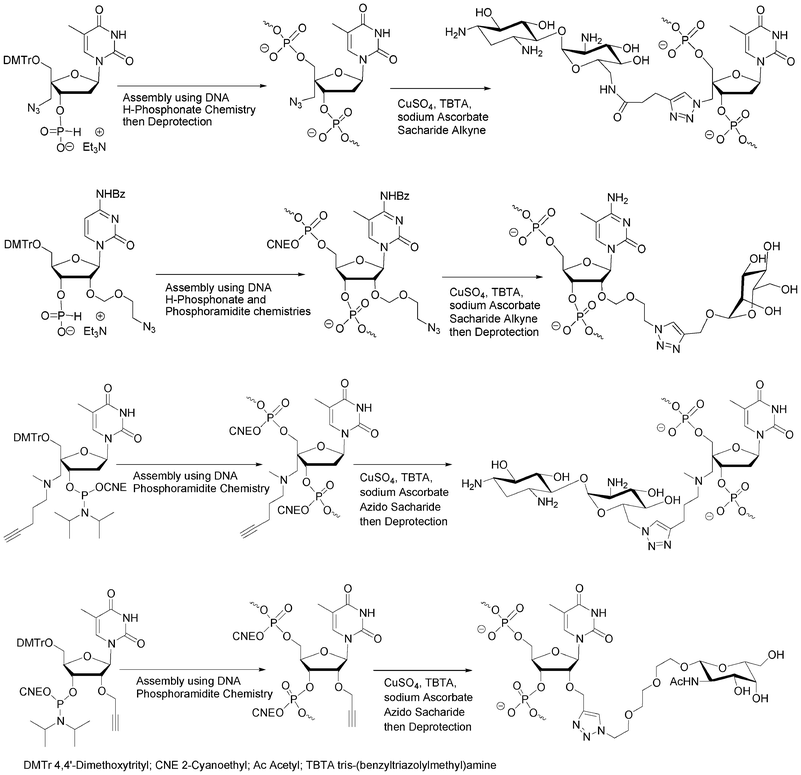 | ||
| Fig. 9 Azido68,69 and alkyne70,71 nucleosidic building blocks to obtain oligonucleotide-saccharide conjugates using CuAAC. | ||
Recently Micura et al. overcame the problem of Staudinger reaction using 2′-deoxy-2′-azido protected cytidine or guanosine phosphodiester building blocks72 which were introduced into the RNA sequence by phosphotriester chemistry73 for a further conjugation with a tetramethylrhodamine alkyne derivative.
In order to allow 5′-end conjugation with azido saccharides close to each other in case of multiconjugation, non-nucleosidic phosphoramidites containing one,74 two and three alkyne functions were prepared.75 (Fig. 10) Monoalkyne 1 and dialkyne phosphoramidites 2 were prepared in one step by reaction of pent-4-yn-1-ol with 2-cyanoethyl N, N, N′, N′-tetraisopropylphosphorodiamidite (in presence of diisopropylammonium tetrazolide) and diisopropylphosphoramidous dichloride in presence of triethylamine, respectively (Scheme 1).
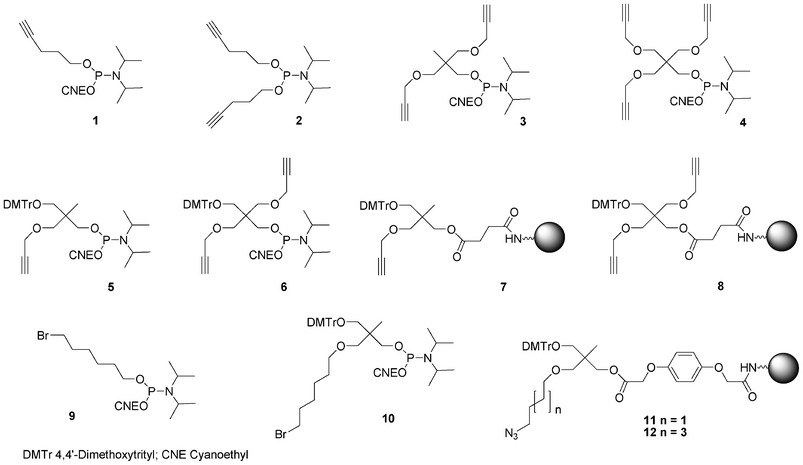 | ||
| Fig. 10 Non-nucleosidic phosphoramidites and solid support building blocks used in the post-conjugation strategy to obtain DNA-like glycomimetics described herein. | ||
 | ||
| Scheme 1 Synthesis of mono and di-pentynyl phosphoramidites. | ||
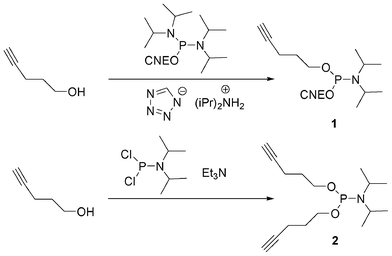
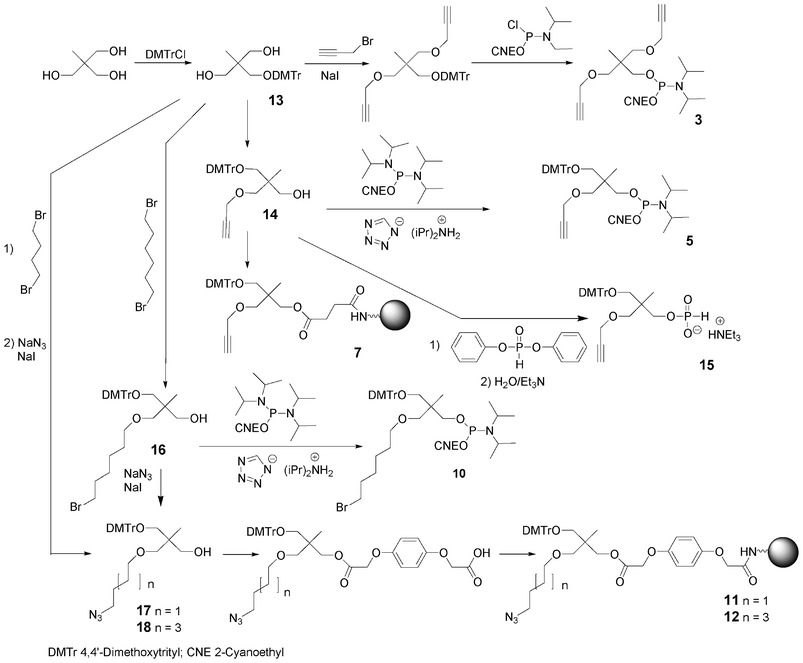
Bis-alkyne derivative 3 was obtained from 2-(hydroxymethyl)-2-methylpropane-1,3-diol. Its dimethoxytritylation to afford 2-(4,4′-dimethoxytrityloxymethyl)-2-methylpropane-1,3-diol 1366 was followed by bis-alkylation with propargyl bromide in the presence of sodium hydride. After removal of the DMTr group in acidic medium, the alcohol was phosphitylated by reaction with diisopropyl cyanoethyl chlorophosphoramidite (Scheme 2). Similarly, the tris-alkyne derivative 4 was prepared from pentaerythritol, except that the intermediate 2,2,2-tris-(propargyloxymethyl)-ethan-1-ol76 was phosphitylated with 2-cyanoethyl tetraisopropylphosphorodiamidite under activation with diisopropylammonium tetrazolide (Scheme 3).
 | ||
| Scheme 2 Synthesis of building blocks from 2-(hydroxymethyl)-2-methylpropane-1,3-diol. | ||
 | ||
| Scheme 3 Synthesis of building blocks from pentaerythritol. | ||
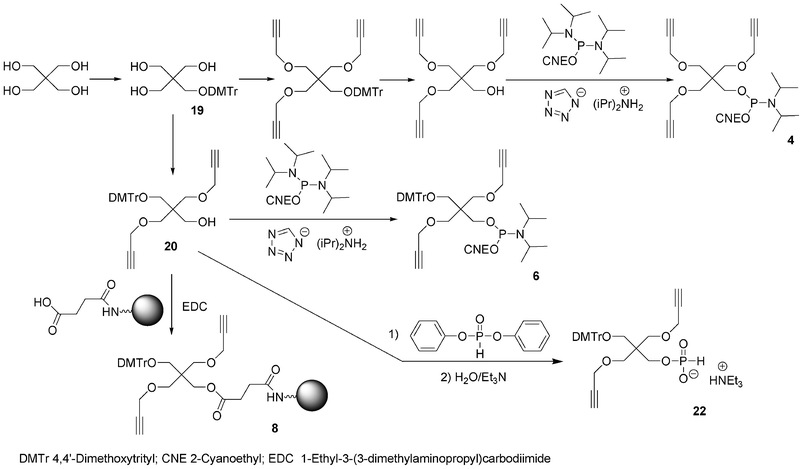
To allow further elongation using DNA chemistry and move the saccharides units away from each other into glycomimetics, two phosphoramidites bearing one alkyne (compound 5, Fig. 10)77 and two alkyne functions (compound 6, Fig. 10) and an hydroxyl function protected as a DMTr ether78 were here again obtained from 2-(hydroxymethyl)-2-methylpropane-1,3-diol and pentaerythritol, respectively. 2-(4,4′-Dimethoxytrityloxymethyl)-2-methylpropane-1,3-diol 13 was monoalkylated with propargyl bromide giving rise to a compound 14 with a free hydroxyl function which was phosphitylated using 2-cyanoethyl tetraisopropyl phosphorodiamidite activated with diisopropylammonium tetrazolide to provide the building block 5 (Scheme 2). Similarly, after dimethoxytritylation of pentaerythritol leading to compound 19, bis-alkylation with propargyl bromide afforded an intermediate 20 with a free hydroxyl function which was phosphitylated to lead to the phosphoramidite 6. (Scheme 3)
In order to enable 3′-end conjugation, the dimethoxytritylated mono- or bis-alkyne intermediates 14 and 20 were loaded on a succinyl LCAA CPG yielding the solid-supported units 7 and 8 bearing one and two alkyne functions, respectively (Schemes 2 and 3).66
Instead of alkyne functions able to react with azido saccharides, Lönnberg59 had reported the use of a phosphoramidite bearing two oxyamines protected with phthaloyl groups able to react with aldehydo saccharides after deprotection (Scheme 4). One hydroxyl function of the phosphoramidite building block was protected as a trityl ether to allow elongation. This phosphoramidite unit and thymidine phosphoramidite were alternatively incorporated 1 to 3 times into an oligonucleotide. At the end of the elongation on solid support, aldehyde-tethered hyaluronan disaccharides were conjugated on solid supports to the deprotected aminooxy functions of the DNA-like scaffold. Glycoconjugates bearing up to six hyaluronan disaccharides linked to the DNA-like construct through oxime bonds were obtained.
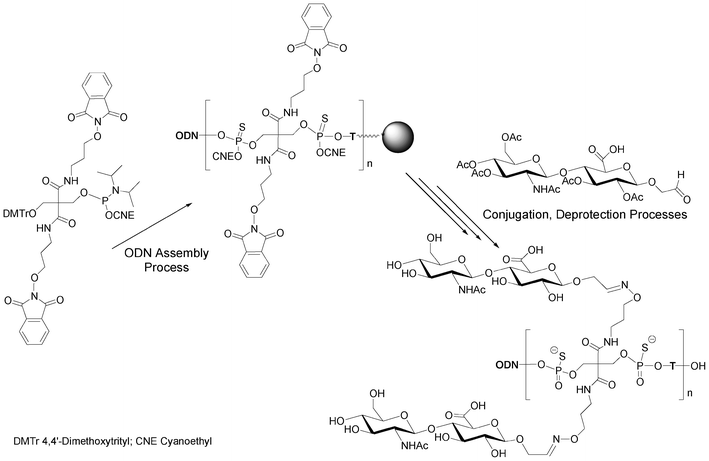 | ||
| Scheme 4 Synthesis of multivalent (n = 1 to 3) hyaluronan disaccharide conjugates of oligonucleotide phosphorothioates59 bearing up to 6 disaccharides. | ||
As mentioned before, due to the reactivity of azides with phosphoramidites, the azide function cannot be introduced in DNA-like scaffolds as a phosphoramidite building block. Instead building blocks containing a bromide atom as a pro-azide function were synthesized. To allow 5′-end conjugation, a bromohexyl phosphoramidite 965,66 was synthesized in one step from 6-bromohexanol and was coupled using standard phosphoramidite elongation. The displacement of the bromine atom to provide the terminal azido group is then carried out on a solid support by reaction with a mixture of sodium azide and sodium iodide in DMF or using tetramethylguanidine azide in acetonitrile. Multiple incorporations of the bromo function into a DNA-like scaffold were performed with the use of phosphoramidite 10 synthesized from 1-(4,4′-dimethoxytrityloxymethyl)-2,2-bis(hydroxylmethyl) propane 13 by monoalkylation with 1,6-dibromohexane and sodium hydride in the presence of sodium iodide followed by phosphitylation using cyanoethyl tetraisopropylphosphorodiamidite activated with diisopropylammonium tetrazolide (Scheme 2).79 Finally to allow 3′-end conjugation through CuAAC, two azido solid supports 11 and 12 were easily prepared from compound 13 by alkylation with either 1,4-dibromobutane or 1,6-dibromohexane in the presence of sodium hydride followed by treatment with a mixture of sodium azide and sodium iodide. The resulting azido compounds 17 and 18 were then transformed into their O-hydroquinone-O,O′-diacetic hemiester derivative (Q-linker)80 which was anchored on LCAA-CPG under EDC activation leading to 11 and 12.74,81
I.4. H-Phosphonate chemistry: more tools in the lego box
H-Phosphonate chemistry is another method to synthesize oligonucleotides on a solid support.82,83 While less popular than the phosphoramidite chemistry, this method is really useful for easily obtaining a variety of oligonucleotide analogues. The DNA synthesis cycle following the H-phosphonate methodology has only two steps (Fig. 11). The detritylation of a nucleoside anchored to the solid support, as for the phosphoramidite cycle, is followed by a coupling step with a H-phosphonate nucleoside. The activation of this coupling step is generally performed with a sterically hindered acid chloride such as pivaloyl or adamantoyl chloride and gives rise to a supported dinucleoside with an H-phosphonate diester linkage. There is no need for a capping step since the acyl chloride is able to react with the 5′-OH functions that have not reacted during coupling. There is also no need for an oxidation step following the coupling step since H-phosphonate diesters are stable during the next detritylation and coupling steps. The oxidation is thus generally performed at the end of the elongation. A diverse range of analogues with modified internucleoside linkages could be thus obtained from the same H-phosphonate diester oligonucleotide. Oxidation with iodine/water gives rise to phosphodiester oligonucleotides (Fig. 11, X = O). Reaction with elemental sulfur affords phosphorothioate internucleoside linkages (Fig. 11, X = S).84 Oxidation with a borane amine complex leads to boranophosphates (Fig. 11, X = BH3).85 Finally, oxidative amidation by carbon tetrachloride in the presence of an amine generates oligonucleotide phosphoramidates (Fig. 11, X = NHR).86–88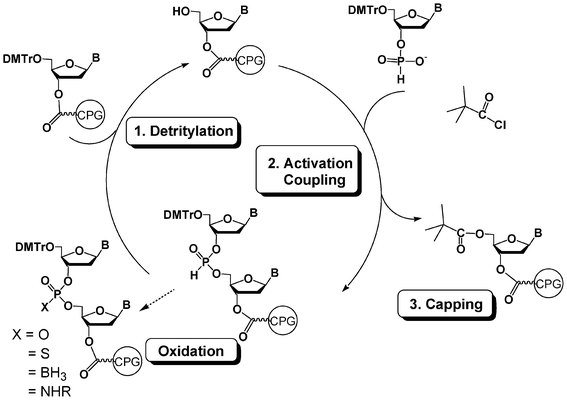 | ||
| Fig. 11 DNA synthesis cycle using H-phosphonate chemistry. | ||
Non nucleosidic H-phosphonate monoester building block analogues of phosphoramidite compounds bearing one or two propargyl groups were synthesized in a one-pot two step reaction from 2-(4,4′-dimethoxytrityloxymethyl)-2-propargyloxymethyl-propanol 14 (Scheme 2) or 2-(4,4′-dimethoxytrityloxymethyl)-2,2-bis-propargyloxymethyl-ethanol 20 (Scheme 3), respectively. Reaction of these compounds with an excess of diphenylphosphite followed by hydrolysis with water and triethylamine lead to the expected H-phosphonate monoester derivatives 15 and 22 respectively.89 The DNA-like scaffolds obtained with these derivatives (vide infra) have the alkyne functions displayed on non-nucleosidic moieties separated from each other by phosphodiester analogue linkages.
Another possibility offered by the H-phosphonate methodology is the introduction of an alkyne or pro-azide function onto the internucleosidic linkage through an oxidative amidation with CCl4 in presence of propargylamine or 3-bromopropylamine, respectively (Scheme 5). In such a strategy, the clickable functions carried by the phosphorus linkages are separated by units acting as linkers. Any organic molecule bearing two alcohol functions could act as a linker. For this purpose we have been using mostly two non-nucleosidic H-phosphonate monoesters 23 and 24 synthesized from cyclohexane-1,4-diyldimethanol90 and tetraethyleneglycol91 (Fig. 12). Dimethoxytritylation and phosphitylation of these diols is performed as for derivatives 15 and 22.
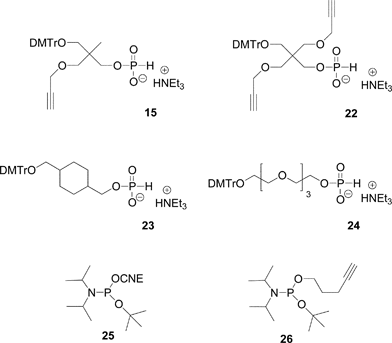 | ||
| Fig. 12 Non-nucleosidic H-phosphonate and phosphoramidite building blocks giving rise to H-phosphonate diesters used in the post-conjugation strategy to obtain DNA-like glycomimetics. | ||
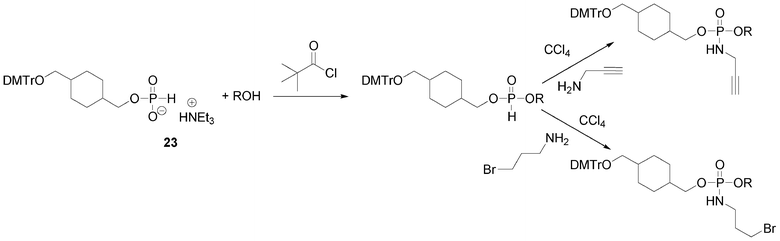 | ||
| Scheme 5 Oxidative amidation to introduce alkyne or bromoalkyl function. | ||
The propargylamino group could also be introduced at the 5′-end of the DNA-like scaffold using a combination of phosphoramidite and H-phosphonate methodologies. Indeed, we and others have shown and used the release in acid medium of benzyl61,92 or tert-butyl93 of corresponding phosphite triesters giving rise to H-phosphonate diesters through an Arbuzov-like reaction. Then subsequent oxidative amidation in presence of propargylamine yields the desired phosphoramidate bearing the alkyne function (Scheme 6). The 5′-O-modification is introduced by means of 2-cyanoethyl tert-butyl N,N-diisopropyl phosphoramidite 25 (Fig. 12)94 simply prepared by reaction of tert-butanol with 2-cyanoethyl tetraisopropylphosphorodiamidite in presence of diisopropylammonium tetrazolide. After its incorporation into an oligonucleotide using phosphoramidite chemistry, the corresponding phosphite triester is not oxidized as in a standard cycle but directly treated with dichloroacetic acid used for the removal of dimethoxytrityl to afford an H-phosphonate diester that could be further oxidized to introduce an alkyne function for subsequent CuAAC conjugation (Scheme 6).
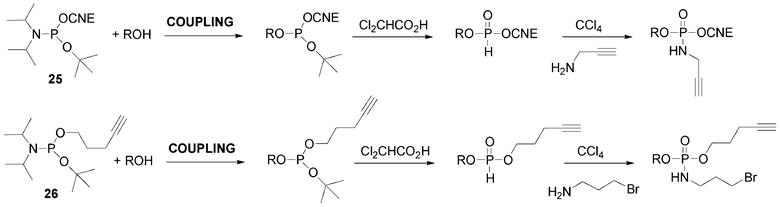 | ||
| Scheme 6 tert-Butyl phosphoramidites to form H-phosphonate diester linkage and further amidative oxidation to introduce alkyne or bromoalkyl groups. | ||
Introduction of both bromoalkyl as precursor of an azide function and alkyne functionalities could be done according to a similar strategy using pent-4-ynyl tert-butyl N,N-diisopropyl phosphoramidite 26 (Scheme 6).95 After coupling at the 5′-position of a nucleoside (ROH), the phosphite triester is treated with dichloroactic acid giving rise to an H-phosphonate diester. Then, an amidative oxidation was performed with 3-bromopropylamine/CCl4 to give a compound with 5′-terminal bromopropyl and pentynyl groups (Scheme 6). Thus the first CuAAC allows the introduction of an azide derivative on the alkyne and the subsequent bromine substitution by NaN3 followed by a second CuAAC with an alkyne derivative leads to hetero-bis-conjugated oligonucleotides.95
I.5. From the tool box to complex molecular architectures
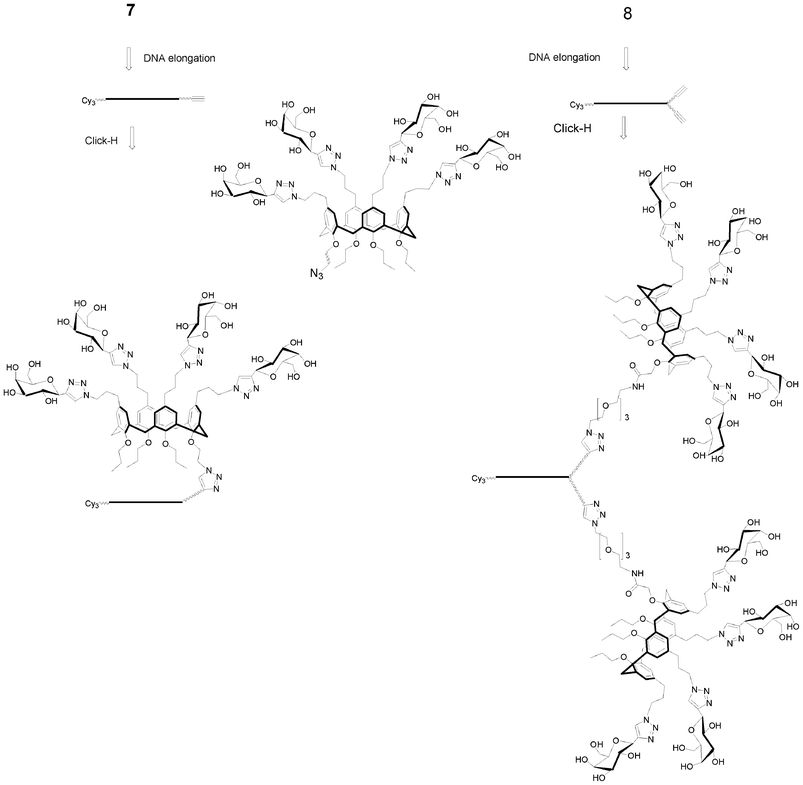
The approach that we have developed for the synthesis of DNA-like glycomimetics falls within the principles of synthetic biology. Indeed, from extremely simple building blocks, molecules of complex architectures mimicking nature and interacting with natural molecules are obtained. As a general rule, we developed strategies to synthesize glycomimetics with different valencies and topologies like linear comb-like or antenna-like structures and also dendrimers on calixarene- or hexapyranose-cores.I.5.1.1. Functionalization of inter phosphodiester linkages: phosphoramidite chemistry. The synthesis of Cy3-DNA glycomimetics is straightforward starting from the alkyne solid support 7, monoalkyne phosphoramidite 5 and commercially available nucleoside phosphoramidites using a DNA synthesizer. All steps are automated affording the solid-supported oligonucleotide polyalkynes which could be conjugated directly onto the support or in solution after deprotection. It is noticed that CuAAC on the solid support is more robust and reliable under microwave assistance while in solution the use of microwaves is less important. In addition conjugation in solution requires more copper than on solid support probably due to some interaction of copper with phosphodiester linkages leading to less copper being available for catalysis.
I.5.1.2. Functionalization on phosphodiester linkages: H-Phosphonate chemistry. Linear glycomimetics are easily accessible by H-phosphonate chemistry and amidative oxidation in presence of propargyl amine to introduce the alkyne function.90,91 Thus different linear glycomimetics, with a DNA tag, exhibiting carbohydrate residues were synthesized from H-phosphonates 23 or 24, amidative oxidation followed by a CuAAC to conjugate the corresponding azide carbohydrate derivatives (galactose, mannose or L-fucose). The resulting linear glycomimetics are neutral (Fig. 13). The glycomimetics constructed from 23 have a semi-rigid hydrophobe linker of 8-atom length and those from 24 have a flexible hydrophilic linker of 13-atom length.
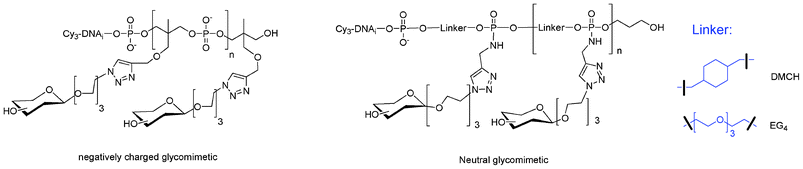
 | ||
| Scheme 7 Solid phase synthesis of antenna-like fucosylated pentaerythrityl phosphodiester oligomers. | ||
The same strategy was applied to synthesize the Cy3-DNA antenna glycomimetic bearing ten galactose residues for binding studies with galactose specific lectins (RCA120 and PA-IL).96
I.5.3.1. Combinatorial synthesis. Combinatorial synthesis is an interesting strategy to synthesize rapidly libraries of molecules that could be tested together to identify a hit through deconvolution of the library. This strategy is less time consuming and allows high throughput screening.
Small libraries of DNA-glycoclusters were synthesized starting from polyalkyne thymidine on which two galactose azides exhibiting different linkers (propyl and dimethylcyclohexane) were conjugated together by co-clicking. Thus a dialkyne led to four different glycoclusters, a trialkyne to eight and a tetraalkyne to sixteen (Fig. 14). HPLC analyses showed a difference of reactivity between both azides leading to a non statistic repartition of the glycoclusters. This proof of concept opens the way to the preparation of more complex libraries.
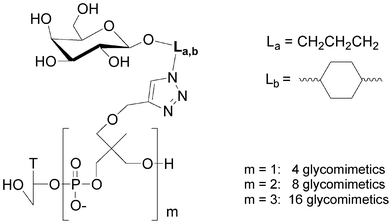 | ||
| Fig. 14 Libraries of galactoclusters synthesised by combinatorial click chemistry. | ||
I.5.3.2. Sequential click chemistry.
I.5.3.2.1 Two sequential CuAAC click chemistries using a bromo derivative as a pro-azide function. Three strategies were reported to synthesize heteroglycoclusters (Scheme 8).
i) Non nucleosidic bromohexyl and propargyl phosphoramidites were coupled alternately to an oligonucleotide by phosphoramidite chemistry leading to a scaffold exhibiting two alkyne and two bromohexyl functions.79 A first click reaction with galactose azide introduced two residues. Then, the two bromine atoms were substituted by sodium azide to reach two azidohexyl groups on which two propargyl mannosides were conjugated by a second click affording a glycocluster with two mannose and two galactose alternate motifs.
ii) Phosphoramidite 26 bearing a pentynyl and a tert-butyl group (Scheme 6) was used to synthesize an oligonucleotide with a triple bond and a H-phosphonate diester linkage that was oxidized in presence of 3-bromo-propylamine affording the 5′-pentynyl bromopropyl phosphoramidate oligonucleotide.95 It was then bis-conjugated according to the first strategy leading to a galactose/mannose cluster displaying the carbohydrate epitopes in close vicinity.
iii) In the third strategy, protected cytidine alkylated on position 2′ with a (2-bromoethoxy)methyl or a (2-azidoethoxy)methyl were converted into their phosphoramidite and H-phosphonate derivatives respectively.68 They were introduced into 2-O-methyl oligonucleotide once mixing phosphoramidite and H-phosphonate chemistries. Propargyl mannoside was introduced by a first click and after bromine substitution by sodium azide, a propargylated galactose was conjugated by a second click affording the oligonucleotide decorated by two different carbohydrates.
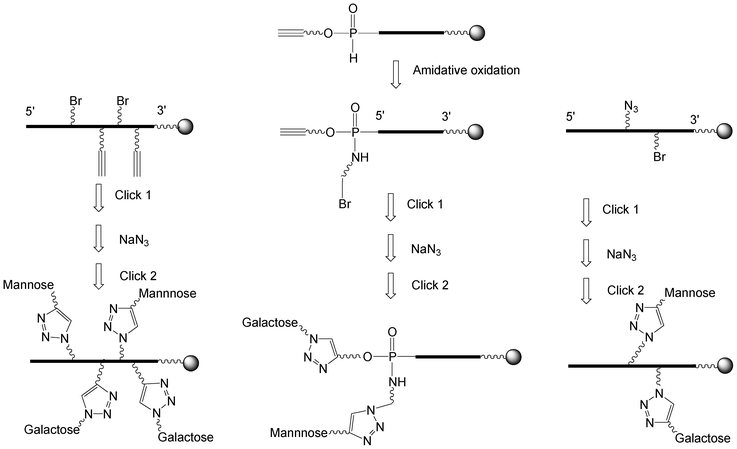 | ||
| Scheme 8 Strategies to synthesize heteroglycoclusters using CuAAC. | ||
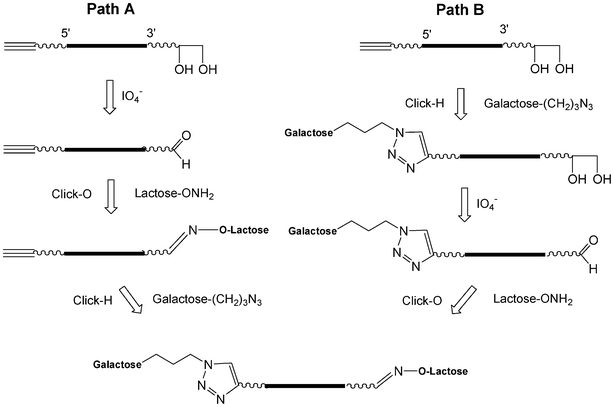
I.5.3.2.2. Sequential CuAAC and an oxime formation (click-H click-O). The CuAAC (click-H) is orthogonal with the oxime formation (click-O).99 Starting from a glycerol solid support, a 5′-pentynyl oligonucleotide was synthesized which after deprotection leads to an oligonucleotide bearing a 3′-diol as the precursor of an aldehyde function. The sequential click reactions could be performed according to two paths (Scheme 9).100 Via path A, the diol was oxidized by NaIO4 and after desalting by size exclusion chromatography (SEC) the click-O reaction was applied using lactose aminooxy working at pH 4.5. After increasing the pH to 7, the click-H reaction was performed with galactose propylazide using CuSO4 and sodium ascorbate. Both click reactions could be performed in one pot. For path B, the click-H reaction was performed first, then the oxidation and finally the click-O reaction. In this case desalting by SEC is required between each step.
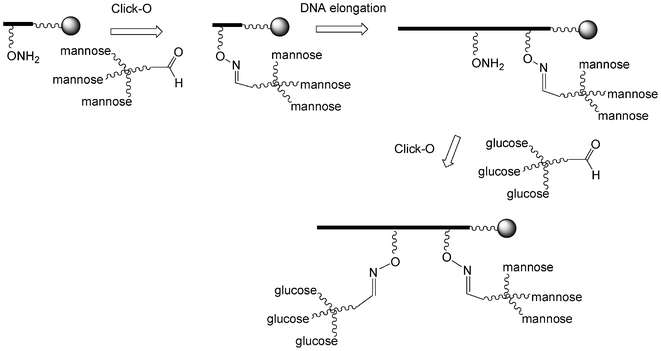
I.5.3.2.3. Two sequential click oxime formations (click-O). Lönnberg et al. reported the use of two sequential click-O reactions to introduce both tri-mannosylated and tri-glucosylated scaffolds into an oligonucleotide (Scheme 10).101 To this end, the trivalent derivatives were first synthesized by click-H reactions using a 6-azido-6-deoxy-1-O-methyl-hexose derivative and tri-propargylpentaerythrityloxy benzaldehyde. Then a phosphoramidite building block with a protected aminooxy function was introduced once and after its deprotection the trimannose aldehyde was conjugated by a click-O reaction. The two steps were reproduced using a tri-glucose aldehyde and the oligonucleotide was elongated. After final deprotection the tri-mannose/tri-glucose oligonucleotide was obtained.
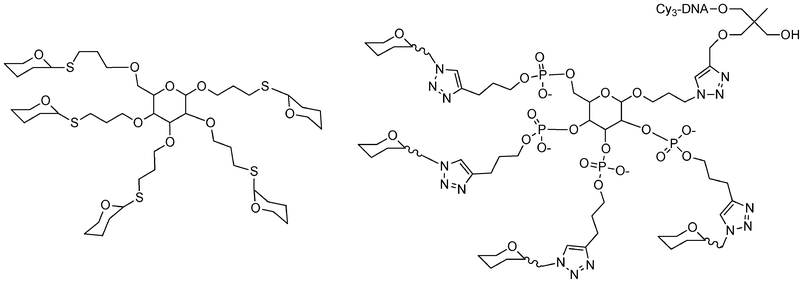 | ||
| Fig. 15 Structure of different carbohydrate-centered glycoclusters synthesized by glycosylation106 or phosphoramidite and click chemistries.81 | ||
I.5.5.1. Linear glycomimetics. To this end, we used the phosphoramidite or H-phosphonate of 1,1,1-tris(hydroxymethyl)ethane THME (5 or 15). Compound 5 leads to DNA-glycomimetics with negatively charged phosphodiester linkages using phosphoramidite chemistry. In contrast, compound 15 leads to DNA-glycomimetics with H-phosphonate diester linkages which are easily transformed into phosphoramidate by amidative oxidation with CCl4 in presence of methoxyethylamine (MOE) or dimethylaminopropylamine (DMAP) leading to the corresponding neutral or positively charged glycomimetic moieties (Fig. 16). In this case it is necessary to use a combination of H-phosphonate and phosphoramidite chemistries108 for the construction of the alkyne scaffold and of the DNA tag respectively. The glycomimetics were labelled with a specific DNA sequence for further multiplexing using a DDI-glycoarray (see below).
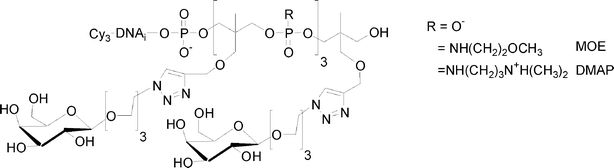 | ||
| Fig. 16 Linear glycomimetics with different electronic environments. | ||
I.5.5.2. Antenna glycomimetics. Like for linear glycomimetics, antenna-like ones were synthesized with a negatively charged phosphodiester or positively charged phosphoramidate linkages, starting from the bis-alkyne solid support 8 which was coupled either with bis-alkyne phosphoramidite 6 or with H-phosphonate 22. The resulting tetraalkyne modified oligonucleotides were conjugated with a galactose azide derivative (Fig. 17).
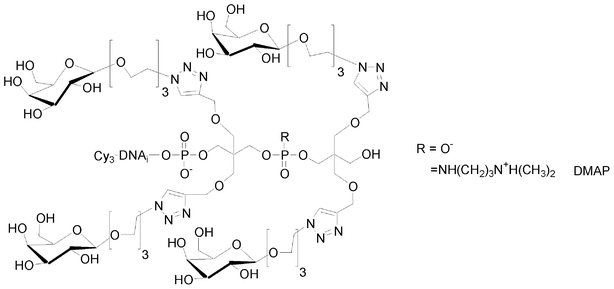 | ||
| Fig. 17 Antenna glycomimetics with different electronic environments. | ||
 | ||
| Scheme 11 Synthesis of calixarene supported galactosyl clusters.96 | ||
II. DNA-based carbohydrate microarrays
II.1. Methods for studying carbohydrate-lectin interactions
To study and understand carbohydrate-lectin interactions, several techniques are available for measuring equilibrium binding constant, interatomic distances, rate of reactions, etc. Roughly, these techniques can be categorized into biophysical methods (X-ray diffraction, mass spectrometry and NMR), equilibrium based methods (equilibrium dialysis, affinity chromatography, affinity co-electrophoresis), kinetic methods (Surface Plasmon Resonance), thermal methods (titration calorimetry), and biochemical methods (enzyme-linked immunosorbent assay-based approaches, precipitation-based assays, hemagglutination-based assays). These methods have been very valuable and have brought key thermodynamic, kinetic and structural parameters.109,110 However, all these techniques have some common drawbacks: they often require large amount of material or are labour intensive or require expensive instruments. Furthermore, some of these techniques cannot cope with the high structural diversity of carbohydrates as they are not high throughput.II.2. Glycoarrays
Glycoarrays or carbohydrate microarrays have emerged over the past two decades as high throughput tools to study protein–carbohydrate interactions.111–130 Carbohydrate microarrays consist of various carbohydrate immobilized (probes) in an array fashion at the surface of a solid material. Therefore it allows the simultaneous detection of multiple interactions with pico- to nanomoles of proteins and carbohydrates. Upon incubation with the targeted organism, protein or biomolecules, interactions with carbohydrates are identified by mapping a measurable physical signal from the substrate surface (e.g. fluorescence, change of refractive index). Some analyses rely on final point analysis, such as fluorescence based microarrays, and some others can perform real time analysis, such as Surface Plasmon Resonance (SPR) based arrays.A key step in the fabrication of a glycoarray is the immobilization of the carbohydrate residues on the surface of the material. Indeed, immobilization of the carbohydrate probe should preserve the key/lock interaction between the probe and the target, have sufficient binding sites available, be homogenous and reproducible. Apart from mass transport considerations, the observed signal at equilibrium is a function of the surface density of the active probe, of the affinity constant and of the target concentration. In binding assays, only the affinity constant is biologically meaningful.131 Therefore, it is important to have a similar surface density for each of the immobilized probes. However, despite the fact that the same reaction is used for the immobilization, reaction yields can be different for each probe. Therefore, quality control of surface bound probe is important.91,132–134
Furthermore, the number of active probes may be influenced by steric hindrance considerations which are related to probe surface density, probe surface organization (in islands for example), probe surface orientation, distance from the surface, or the spacer’s chemical nature.
Immobilization of carbohydrates on solid support (substrate) has been achieved through physisorption, chemisorption or through biological interactions.
Physisorption relies on weak interactions between the carbohydrate probe and the substrate. Its main advantage is simplicity but leaching of adsorbed molecules is an issue. While polysaccharides can be immobilized without further modifications, low molecular weight carbohydrates require the introduction of an anchoring tail such as lipid tails,117,135 fluorescent tags,136 proteins122 or polymers122 to enhance the interaction with the substrate.
Carbohydrates can be immobilized covalently without modification through their reductive end137 or by derivatization with an aglycon that carries a reactive chemical entity for surface coupling. Surface coupling of the glycoside can be achieved directly without surface chemical modification or can be achieved after introduction of new chemical function at the surface of the material (surface chemical functionalization). For example, mercaptan glycosides can be reacted directly on gold138 or with maleimido modified surfaces.125
Covalent immobilization has been reported by reacting thiols with double bonds or other thiols, but also amines (or hydrazine) with activated esters,126,139,140 cyanuric acid chlorides,141 aldehydes142 or epoxides.143 Ligation reactions such as Staudinger144 or cycloaddition118,145–147 have also been reported.
Finally, carbohydrate immobilizations have been achieved through biochemical interaction or reactions. Avidin/biotin interactions have been widely reported.148–150 In this case, carbohydrates are derivatized with a biotin and then allowed to react with an avidin layer. Another interaction reported for the immobilization of carbohydrates is the immobilization of the DNA-carbohydrate conjugates in a highly specific manner onto a DNA microarray through Watson–Crick hybridization. It was already shown that covalent modification of the 5′-terminus of DNA with carbohydrates did not interfere with the hybridization of DNA glycoconjugates with complementary DNA.151 In the following we will discuss of this latest immobilization technique which is also called DNA directed immobilization (DDI).
II.3. DDI and DNA-based glycomimetics
In 2010, Niemeyer reviewed the synthesis and applications of DNA conjugates152 such as in bioanalytics and in nanofabrication. In bioanalytic applications, DNA-conjugates have been exploited for the fabrication of biochips (microarrays), particularly for the immobilization of proteins. Each DNA-conjugate is selectively addressed and immobilized at a specific location of the array where its complementary DNA sequence is immobilized. The selectivity of the Watson–Crick hybridization can be considered as a molecular actuator for sorting molecules at the surface of the array according to their complementary sequences. PNA based DDI arrays have also been reported.153,154Advantages of DDI are mild chemical immobilization conditions and long term stability of the DNA platform. The DNA microarray platform is reusable upon regeneration using alkaline solution. DDI microarrays are compatible with various transduction techniques such as mass spectrometry,155 fluorescence91 or electrochemical impedance.156 Furthermore, DDI allows the interaction between the probe and the target to be conducted in solution before addressing the whole complex on the DNA array thanks to the DNA tag22,91,155,156 and will be called the “in-solution” approach in the following (Fig. 18).
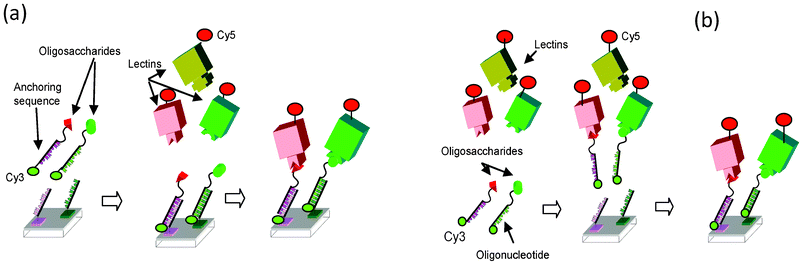 | ||
| Fig. 18 General schematic of DDI based glycoarray. a) “On-chip” approach: glycomimetics are first immobilized by hybridization on DNA array, next the lectins are allowed to interact with immobilized glycomimetics. b) “In-solution” approach: after interaction in solution of the glycomimetics with the lectins, the complexes are fixed on the DNA array by hybridization. Cy3 is related to the glycomimetic surface density, and Cy5 (or Alexa 647) are related to the lectins. Reproduced from Chevolot et al.91 with the permission of Wiley. | ||
Finally, Wacker et al. have compared the signal intensity, limit of detection and reproducibility of immuno-assays using either covalently or DDI immobilized antibodies.157 DDI and covalent immobilization led to similar signals. However, in the case of DDI, a 100-fold less antibody concentration was required and better reproducibility was observed.157 The reason for this difference was attributed either to a better availability of DDI immobilized probe or a better conformational freedom in the case of DD1.152,157
DDI arrays have been employed to study small molecule/protein interactions,154 aptamer/antibody interactions,156 peptide/cell interactions158 or to discover new proteolytic activities in dust mite extract.153 In the following, we will focus on glycomimetic DDI arrays and their use to study lectin–glycomimetic interactions.
Interaction of covalently- and DDI-immobilized glycomimetics with Ricinus communis agglutinin 120 (RCA 120, a galactose specific lectin) will be discussed first. Next, quantitative evaluation of glycomimetic–lectin interactions will be described followed by the description of the “in-solution” approach. Finally, the development of a miniaturized multiplexed DDI glycomimetic array will be presented. In our work, DNA-glycomimetics are labelled with a Cyanine 3 fluorescent dye (Cy3). The targeted lectins are labelled with a Cyanine 5 or Alexa 647 dye. This dual fluorescence read-out allows control of the relative surface immobilization of the glycomimetics (Cy3) and study of the lectin–glycomimetic interactions (Cy5).
In both case, the Cy5 signal was found to be higher when the galactomimetic was immobilized by DDI, suggesting that a higher level of DDI immobilized galactomimetics interacted with RCA 120. However, the difference in Cy5 signal was more significant for the lowest concentration. This is in agreement with the results of other groups.157
Several groups have reported the measurements of competitive inhibition experiments (IC50)124 or dissociation constant (Kd)160,161 for carbohydrate–lectin interactions using a fluorescence read-out based on microarray technology.
These assays were adapted to DDI glycomimetic arrays. Classically in IC50 experiments, the studied molecule is considered as the inhibitor of the interaction of interest — let’s say between a protein and its natural ligand. The assay consists of incubating the protein with its natural ligand at a given concentration with increasing concentrations of the inhibitor of interest. Therefore, low IC50 values are considered to illustrate a high affinity. In our assay, after the immobilization of the glycoconjugates of interest at the bottom of each microreactor, each microwell is individually incubated with different mixtures of increasing concentration of inhibitor (e.g. lactose) and lectin (e.g. RCA 120, PA-IL) at the desired concentration. The IC50 is determined as the concentration of inhibitor (lactose) required to reduce the fluorescence signal of the labelled lectin (Cy5 or Alexa 647) by 50%. The higher the concentration of inhibitor required to achieve 50% of inhibition, the better the affinity of the lectin for the glycomimetic bound at the surface of the microarray. This competition assay uses picomoles of glycomimetic and lectin while conventional methods (ELLA or ELISA) requires 20000-fold more of materials.
We found that the relative potencies per carbohydrate residue as determined by direct fluorescence scanning and by IC50 measurements were different, underlying the importance of complete single affinity threshold-based assays with regard to semi-quantitative or quantitative assays.
The concept was demonstrated using two lectins RCA 120 (a galactose binding lectin) and PA-IIL (a fucose binding lectin) in combination with two glycomimetics bearing two different DNA sequences (a galactosylated glycomimetic and a fucosylated glycomimetic).162 The two complementary sequences of the two glycomimetics were covalently immobilized in alternate lines of eight spots on a DNA microarray.
It was demonstrated that glycomimetics were specifically addressed according to their sequences. When the galactosylated glycomimetic and the fucosylated glycomimetic labelled with Cy3 were incubated together with Cy5-RCA 120, the Cy3 fluorescence signal was similar on all lines, illustrating that both glycoconjugates were correctly addressed, but fluorescence at 635 nm (Cy5) was only observed for the lines corresponding to the DNA sequence complementary to the one carried by the galactosylated molecule. This result demonstrates that the RCA 120 lectin specifically recognized the galactomimetic from the mixture and that the lectin-glycoconjugate complex was efficiently addressed at the desired spot in the microwell due to the specificity of DNA/DNA hybridization. A similar result was obtained with PA-IIL, where the Alexa 647 fluorescence signal was only observed for the lines corresponding to the DNA sequence complementary to the one displaying fucosylated molecules.
These results showed that the two specific recognitions (RCA 120/Gal and PA-IIL/Fuc) were successfully performed and the resulting complexes were specifically addressed on the DNA chips. The DDI array combines both advantages of the recognition in solution (no artefact due to the vicinity of the surface) and the advantages of microarrays (multiplexing with low amounts of material).
The designed microdevice was a glass slide bearing 40 microwells at the bottom of which 64 to 144 spots could be printed (Fig. 19). Thanks to these microwells, several experiments with varying experimental conditions could be carried out simultaneously on the same glass slide. For example, each microwell corresponds to one specific concentration of inhibitor (IC50) or of lectin (saturation curve) allowing for the simultaneous determination of the IC50 or Kd of 7 to 10 glycomimetics with only picomoles of each molecule.163
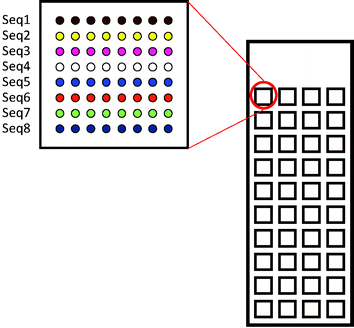 | ||
| Fig. 19 40 square (3 × 3 × 0.065 mm) microreactors are fabricated on microscope borosilicate glass slides. At the bottom of each microwell, 8 to 12 lines of 8 to 12 spots can be printed in successive lines. | ||
For the fabrication of microwell structured glass slides, we have developed a deep etching technology of soda-lime/borosilicate glass based on microtechnology processes and wet or dry etching.164,165 Our process is based on a photoresist/chromium bilayer and on an HCl/buffered oxide etchant mixture.
The design of the DNA anchoring sequences was done from a set of experimental sequences. From these sequences, new sequences were generated in silico by randomly mixing trimer segments. For each sequence, the Gibbs free energies (ΔG) of hybridization with all complementary sequences were calculated by various bioinformatic tools166,167 generating a square matrix. Sequences having similar hybridization ΔG (with its complementary sequence) and cross hybridization ΔGs lower than −6 kcal mol−1 were selected. From 120 calculated sequences, a set of 14 sequences was tested experimentally from which 11 were selected.
Using this multiplex microsystem, six galactoconjugates displaying different charges, linkers and topologies were screened for their binding properties toward PA-IL. This system allowed the determination, in a single experiment the binding of 6 glycoconjugates for PA-IL in solution thanks to the specificity of DNA/DNA duplex formation. With the same platform, their IC50 and Kd values could be simultaneously determined.163
III. Towards the rational design of DNA-based glycomimetics
Our aim was to develop a rapid and flexible synthetic strategy for the generation of a wide variety of glycomimetics structures. These molecules were synthesized at a nanomole scale and screened for binding towards the targeted lectin on the DDI glycomimetic array. Targeted applications were the intention of the design of high affinity glycomimetics with respect to PA-IL and PA-IIL in the hope that such molecules may be applied to prevent pseudomonas aeruginosa adhesion in the respiratory tract of patients (in particular cystic fibrosis patients). RCA120 is a plant lectin chosen as the model lectin, commercially available and well-documented in the literature.Pseudomonas aeruginosa (PA) is the main cause of the morbidity and mortality of cystic fibrosis patients and is the second most frequent bacterium involved in nosocomial infections. PA-IL and PA-IIL are primary virulence factors and cytotoxins of PA. They are also broad-spectrum agglutinins, which may cause bacteria to adhere to host cells leading to respiratory epithelial damage during PA respiratory infections. Despite heavy conventional antibiotic therapy, Pseudomonas aeruginosa infections often lead to chronic pneumonia and eventually to death. Therefore there is an urgent need to develop new therapies. PA-IL and PA-IIL have been described as potential targets for anti-adhesive therapies. Hereafter, our aim was to design high affinity ligands for PA-IL and PA-IIL.
PA-IL and RCA 120 are both galactose binding lectins. RCA 120 is composed of two ricin-like heterodimers linked through weak interactions. The heterodimer consists of an A chain inhibiting protein synthesis in eukaryotic cells and a B chain that has two β-independent galactoside binding domains.168–170 The A and B chains are linked by a single disulfur bridge. PA-IL is tetravalent lectin. It has a nearly rectangular shape with binding sites distances of 71 Å on the long side, and 32 Å on the short side. PA-IIL is a tetravalent fucose binding lectin. It is more or less a two tilted cobblestone-shaped protein leading to a tetrahedral spatial distribution with a distance of 40 Å between the two carbohydrate binding sites.
Carbohydrate-lectin interactions are generally weak (in the μM to mM range) due to the shallow binding site of lectins with a half time of the carbohydrate–lectin complexes in the range of a few seconds.171 An improvement of the binding (decreasing of the Gibbs free energy) can be achieved by three means:
•by stabilizing the complex through additional interactions (decreasing the enthalpy). This approach is efficient if the binding pocket is well-structured,171
•by reducing the entropic cost or by increasing the entropy upon binding. Entropic cost can be reduced by pre-arranging the ligand in its active conformation prior to its interaction with the lectin,171
•by designing multivalent ligands17 to take advantage of the so-called cluster effect.18,19
The weakness of monovalent interactions between lectin and carbohydrate can be compensated by multivalent ligands. Indeed, lectins are usually multivalent. Providing that the topology of the carbohydrate residues of the glycocluster matches the lectin binding sites, the resulting multivalent interaction can be superior to the sum of the individual interactions.13,19 A maximal cluster effect is observed if the multivalent ligand fits at the atomic scale into the different binding sites. However, practically this can not be achieved. Therefore a level of flexibility should be introduced. Consequently, the multivalent interaction of a glycocluster with lectins occurs with an entropic cost. Nevertheless, due to the lack of reliable predictive molecular simulations, the multivalent ligands are often designed through an empirical approach.172
The DNA-based glycomimetics are multivalent ligands and therefore expected to take advantage of the cluster effect providing that their topology matches the one of the targeted lectins. Furthermore, taking advantage of the “chemical toolbox” developed the scaffold or the linker between the carbohydrate residue and the oligonucleotide can be designed so to stabilize the complex through additional interactions.
The affinities of galactosylated clusters with different multivalencies (1, 3, 4, 8 and 10), various spatial arrangements (comb-like, carbohydrate-centered clusters, crown geometry and antenna), charged (negative, neutral or positive) and different linkers (dimethanolcyclohexane DMCH, triethyleneglycol) were tested for their binding with respect to RCA 120 and PA-IL lectins. Direct fluorescence scanning, IC50 and Kd measurements were performed.
For RCA 120, a cluster effect was observed with comb-like structures bearing linker DMCH with a 23-fold increase in potency per galactose residue for trivalent clusters compared to monovalent clusters. At pH 7.4, no charge effect of the glycocluster was observed. The isoelectric point of RCA 120 is between 7.5–7.9. Therefore, it is neutral and should not be affected by the charge of the glycomimetic.
The affinities of galactomimetics bearing one to three positive or negative charges toward PA-IL were probed. Our results suggested that the affinity of PA-IL was enhanced with increased positive charge while increasing negative charge was unfavourable. These observations may be due to attractive or repulsive electrostatic interactions between the lectin and the galactomimetic moiety. Indeed, PA-IL is negatively charged at pH 7.4 (pI = 4.94). IC50 assays suggested that a cationic linker (3+) increased the affinity by 2 orders of magnitude when compared to the same neutral glycomimetic.
For neutral galactomimetics, a comb-like structure was generally preferred and the affinity was enhanced with the DMCH linker. Molecular modelling of the different topologies showed no clear explanations for the observed differences. It may relate to either the hydrophilic-hydrophobic balance of the molecules or to entropic considerations. Indeed, the PA-IL binding pocket displays hydrophobic amino acid residues leading to enhanced affinity for nitrophenyl galactoside compared to galactose. In fact, the affinity of carbohydrate-centered cluster with aromatic linkers was 3 orders of magnitude higher than the same centered structure with propyl, diethyleneglycol or triethyleneglycol linkers. PA-IL showed no affinity for calixarene-based structures. These results were different from those observed by Cecioni et al. who reported a high affinity calix[4]arene-based galactocluster.173 This difference may be related to the fact that in the case of Moni et al., a C-glycoside was used.96 Furthermore, the triazole ring was directly linked to the carbohydrate residue and a 3 carbon linker between the triazole and the calix[4]arene was used. This may have resulted in steric hindrance prohibiting the interaction of the lectin with the galactosyl residues. The bindings of RCA120 and PA-IL to galactomimetics were compared and huge differences in binding were observed. Hence, the binding profiles can be envisioned as a finger print which may be exploited later for sensing applications.
Finally, 16 fucosylated glycoclusters were synthesized and their bindings towards PA-IIL were evaluated on a DDI glycoarray.174 According to our results, monofucosylated derivatives displayed a lower binding affinity for PA-IIL than all other multivalent fucomimetics illustrating the local concentration effect of glycoclusters on binding. The best binding properties were observed for the antenna fucomimetic bearing 4 residues and for the mannose-centered fucomimetics bearing 4 and 8 fucosyl residues. Among the carbohydrate-centered fucomimetics exhibiting 4 residues, the mannose core showed the best binding and a linker effect was observed. An increase in binding was correlated with an increase of the linker length. The linear fucomimetics showed an increase of binding correlated with the number of residues. However, our results suggested that no chelate effect was observed despite the fact that some ligands were able to interact with three binding sites within the same lectin.
Conclusion
The DNA chemistry (phosphoramidite as well as H-phosphonate) in combination with the very efficient copper(I) catalyzed azide alkyne cycloaddition allows the rapid access to various glycomimetics exhibiting different topologies: linear, antenna-like, crown-like and a number of carbohydrate residues. Thanks to a DNA tag, the resulting glycomimetics are easily immobilised on a DNA microarray by specific double-strand formations allowing the multiplexing of analyses. This miniaturized microsystem uses only few tens of picomoles of DNA-glycomimetics for a whole evaluation of their lectin binding properties. This evaluation is performed by direct fluorescence scanning for qualitative information while determination of the IC50 and KD values gives quantitative information. The studies allowed the determination of the structural parameters of glycomimetics important for gaining high-affinities to a specific lectin.Acknowledgements
We thank Dr C. Bouillon, Dr S. Cecioni, Dr X. Chen, Dr J. P. Cloarec, Pr A. Dondoni, T. Géhin, Dr B. Gerland, A. Goudot, Pr M. Phaner Goutorbe, Dr A. Imberty, Dr J. Lietard, C. Ligeour, Pr A. Marra, A. Meyer, Dr L. Moni, Dr G. Pourceau, Dr J. P. Praly, S. Rouanet, A. Thiam, Dr O. Vidal and Dr J. Zhang who greatly contributed to the success of this project. This work was financially supported by the CNRS interdisciplinary program “Interface Physique Chimie Biologie: Soutien à la prise de risque”, ANR-08-BLAN-0114-01, Lyon Biopole, Vaincre la Mucoviscidose, Plateforme NanoLyon, the CNRS and the Région Languedoc-Roussillon. F. M. is from Inserm.References
- R. A. Laine, Glycobiology, 1994, 4, 759–767 CrossRef CAS.
- P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046–1051 CrossRef CAS.
- D. B. Werz, R. Ranzinger, S. Herget, A. Adibekian, C. W. von der Lieth and P. H. Seeberger, ACS Chem. Biol., 2007, 2, 685–691 CrossRef CAS.
- A. Adibekian, P. Stallforth, M. L. Hecht, D. B. Werz, P. Gagneux and P. H. Seeberger, Chem. Sci., 2011, 2, 337–344 RSC.
- G. J. L. Bernardes, B. Castagner and P. H. Seeberger, ACS Chem. Biol., 2009, 4, 703–713 CrossRef CAS.
- O. J. Plante, E. R. Palmacci and P. H. Seeberger, Science, 2001, 291, 1523–1527 CrossRef CAS.
- P. Stallforth, B. Lepenies, A. Adibekian and P. H. Seeberger, J. Med. Chem., 2009, 52, 5561–5577 CrossRef CAS.
- H. J. Gabius, H. C. Siebert, S. Andre, J. Jimenez-Barbero and H. Rudiger, ChemBioChem, 2004, 5, 740–764 CrossRef CAS.
- H. Lis and N. Sharon, Chem. Rev., 1998, 98, 637–674 CrossRef CAS.
- R. J. Pieters, Org. Biomol. Chem., 2009, 7, 2013–2025 CAS.
- G. Cioci, E. P. Mitchell, C. Gautier, M. Wimmerova, D. Sudakevitz, S. Perez, N. Gilboa-Garber and A. Imberty, FEBS Lett., 2003, 555, 297–301 CrossRef CAS.
- A. Imberty, M. Wimmerova, E. P. Mitchell and N. Gilboa-Garber, Microbes Infect., 2004, 6, 221–228 CrossRef CAS.
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2755–2794 CrossRef CAS.
- S. L. Flitsch, Curr. Opin. Chem. Biol., 2000, 4, 619–625 CrossRef CAS.
- J. M. De la Fuente and S. Penades, Biochim. Biophys. Acta, Gen. Subj., 2006, 1760, 636–651 CrossRef CAS.
- A. Imberty, Y. M. Chabre and R. Roy, Chem.–Eur. J., 2008, 14, 7490–7499 CrossRef CAS.
- Y. M. Chabre and R. Roy, in Advances in Carbohydrate Chemistry and Biochemistry, 2010, vol. 63, pp. 165-393 Search PubMed.
- R. T. Lee and Y. C. Lee, in Enhanced biochemical affinities of multivalent neoglycoconjugates, ed. R. T Lee and Y. C. Lee, Academic Press, San Diego, 1994, pp. 23-50 Search PubMed.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS.
- Y. Chevolot, S. Vidal, E. Laurenceau, F. Morvan, J.-J. Vasseur and E. Souteyrand, in Recognition Receptors in Biosensors, ed. M. Zourob, Springer, 2009 Search PubMed.
- Y. Chevolot, ed., Protocols in Carbohydrate Microarrays, Springer verlag, Clifton, 2012 Search PubMed.
- F. Morvan, Y. Chevolot, J. Zhang, A. Meyer, S. Vidal, J.-P. Praly, J.-J. Vasseur and E. Souteyrand, Methods Mol. Biol., 2012, 808, 195–219 CAS.
- S. L. Beaucage and M. H. Caruthers, Tetrahedron Lett., 1981, 22, 1859–1862 CrossRef CAS.
- M. D. Matteucci and M. H. Caruthers, J. Am. Chem. Soc., 1981, 103, 3185–3191 CrossRef CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149 CrossRef CAS.
- H. Lonnberg, Curr. Org. Synth., 2009, 6, 400–425 CrossRef CAS.
- H. Lonnberg, Bioconjugate Chem., 2009, 20, 1065–1094 CrossRef CAS.
- K. Matsuura, M. Hibino, Y. Yamada and K. Kobayashi, J. Am. Chem. Soc., 2001, 123, 357–358 CrossRef CAS.
- K. Matsuura, M. Hibino, T. Ikeda, Y. Yamada and K. Kobayashi, Chem.–Eur. J., 2004, 10, 352–359 CrossRef CAS.
- M. K. Schlegel, J. Hutter, M. Eriksson, B. Lepenies and P. H. Seeberger, ChemBioChem, 2011, 12, 2791–2800 CrossRef CAS.
- K. Gorska, K. T. Huang, O. Chaloin and N. Winssinger, Angew. Chem., Int. Ed., 2009, 48, 7695–7700 CrossRef CAS.
- M. Ciobanu, K. T. Huang, J. P. Daguer, S. Barluenga, O. Chaloin, E. Schaeffer, C. G. Mueller, D. A. Mitchell and N. Winssinger, Chem. Commun., 2011, 47, 9321–9323 RSC.
- C. Scheibe and O. Seitz, Pure Appl. Chem., 2011, 84, 77–85 CrossRef.
- C. Scheibe, A. Bujotzek, J. Dernedde, M. Weber and O. Seitz, Chem. Sci., 2011, 2, 770–775 RSC.
- S. Akhtar, A. Routledge, R. Patel and J. M. Gardiner, Tetrahedron Lett., 1995, 36, 7333–7336 CrossRef CAS.
- T. L. Sheppard, C. H. Wong and G. F. Joyce, Angew. Chem., Int. Ed., 2000, 39, 3660–3663 CrossRef CAS.
- M. Dubber and J. M. J. Frechet, Bioconjugate Chem., 2003, 14, 239–246 CrossRef CAS.
- B. Ugarte-Uribe, S. Perez-Rentero, R. Lucas, A. Avino, J. J. Reina, I. Alkorta, R. Eritja and J. C. Morales, Bioconjugate Chem., 2010, 21, 1280–1287 CrossRef CAS.
- J. Katajisto, P. Heinonen and H. Lonnberg, J. Org. Chem., 2004, 69, 7609–7615 CrossRef CAS.
- M. Adinolfi, L. De Napoli, G. Di Fabio, A. Iadonisi, D. Montesarchio and G. Piccialli, Tetrahedron, 2002, 58, 6697–6704 CrossRef CAS.
- G. Difabio, A. Randazzo, J. D'Onofrio, C. Ausin, E. Pedroso, A. Grandas, L. De Napoli and D. Montesarchio, J. Org. Chem., 2006, 71, 3395–3408 CrossRef CAS.
- E. Alazzouzi, N. Escaja, A. Grandas and E. Pedroso, Angew. Chem., Int. Ed. Engl., 1997, 36, 1506–1508 CrossRef CAS.
- M. Adinolfi, L. De Napoli, G. Di Fabio, A. Iadonisi and D. Montesarchio, Org. Biomol. Chem., 2004, 2, 1879–1886 CAS.
- K. Ketomaki and P. Virta, Bioconjugate Chem., 2008, 19, 766–777 CrossRef.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS.
- V. Aragao-Leoneti, V. L. Campo, A. S. Gomes, R. A. Field and I. Carvalho, Tetrahedron, 2010, 66, 9475–9492 CrossRef CAS.
- A. Dondoni, Chem.–Asian J., 2007, 2, 700–708 CrossRef CAS.
- H. Isobe, K. Cho, N. Solin, D. B. Werz, P. H. Seeberger and E. Nakamura, Org. Lett., 2007, 9, 4611–4614 CrossRef CAS.
- S. Slavin, J. Burns, D. M. Haddleton and C. R. Becer, Eur. Polym. J., 2011, 47, 435–446 CrossRef CAS.
- H. Salo, P. Virta, H. Hakala, T. P. Prakash, A. M. Kawasaki, M. Manoharan and H. Lonnberg, Bioconjugate Chem., 1999, 10, 815–823 CrossRef CAS.
- O. P. Edupuganti, Y. Singh, E. Defrancq and P. Dumy, Chem.–Eur. J., 2004, 10, 5988–5995 CrossRef CAS.
- Y. Singh, O. Renaudet, E. Defrancq and P. Dumy, Org. Lett., 2005, 7, 1359–1362 CrossRef CAS.
- J. Katajisto, P. Virta and H. Lonnberg, Bioconjugate Chem., 2004, 15, 890–896 CrossRef CAS.
- N. Spinelli, O. P. Edupuganti, E. Defrancq and P. Dumy, Org. Lett., 2007, 9, 219–222 CrossRef CAS.
- M. Karskela, P. Virta, M. Malinen, A. Urtti and H. Lonnberg, Bioconjugate Chem., 2008, 19, 2549–2558 CrossRef CAS.
- T. Wada, A. Mochizuki, S. Higashiya, H. Tsuruoka, S. Kawahara, M. Ishikawa and M. Sekine, Tetrahedron Lett., 2001, 42, 9215–9219 CrossRef CAS.
- J. Nielsen and M. H. Caruthers, J. Am. Chem. Soc., 1988, 110, 6275–6276 CrossRef CAS.
- A. M. Jawalekar, N. Meeuwenoord, J. G. O. Cremers, H. S. Overkleeft, G. A. van der Marel, F. Rutjes and F. L. van Delft, J. Org. Chem., 2008, 73, 287–290 CrossRef CAS.
- G. P. Miller and E. T. Kool, Org. Lett., 2002, 4, 3599–3601 CrossRef CAS.
- G. P. Miller and E. T. Kool, J. Org. Chem., 2004, 69, 2404–2410 CrossRef CAS.
- J. Lietard, A. Meyer, J. J. Vasseur and F. Morvan, Tetrahedron Lett., 2007, 48, 8795–8798 CrossRef CAS.
- J. Lietard, A. Meyer, J. J. Vasseur and F. Morvan, J. Org. Chem., 2008, 73, 191–200 CrossRef CAS.
- C. Coppola, L. Simeone, L. De Napoli and D. Montesarchio, Eur. J. Org. Chem., 2011, 1155–1165 CrossRef CAS.
- A. Kiviniemi, P. Virta, M. S. Drenichev, S. N. Mikhailov and H. Lonnberg, Bioconjugate Chem., 2011, 22, 1249–1255 CrossRef CAS.
- A. Kiviniemi, P. Virta and H. Lonnberg, Bioconjugate Chem., 2008, 19, 1726–1734 CrossRef CAS.
- A. Kiviniemi, P. Virta and H. Lonnberg, Bioconjugate Chem., 2010, 21, 1890–1901 CrossRef CAS.
- T. Yamada, C. G. Peng, S. Matsuda, H. Addepalli, K. N. Jayaprakash, M. R. Alam, K. Mills, M. A. Maier, K. Charisse, M. Sekine, M. Manoharan and K. G. Rajeev, J. Org. Chem., 2011, 76, 1198–1211 CrossRef CAS.
- K. Fauster, M. Hartl, T. Santner, M. Aigner, C. Kreutz, K. Bister, E. Ennifar and R. Micura, ACS Chem. Biol., 2012, 7, 581–589 CrossRef CAS.
- J. F. M. de Rooij, G. Wille-Hazeleger, P. H. van Deursen, J. Serdijn and J. H. van Boom, Recl. Trav. Chim. Pays-Bas, 1979, 98, 537–548 CrossRef CAS.
- G. Pourceau, A. Meyer, J. J. Vasseur and F. Morvan, J. Org. Chem., 2009, 74, 6837–6842 CrossRef CAS.
- C. Ligeour, A. Meyer, J. J. Vasseur and F. Morvan, Eur. J. Org. Chem., 2012, 1851–1856 CrossRef CAS.
- A. Mollard and I. Zharov, Inorg. Chem., 2006, 45, 10172–10179 CrossRef CAS.
- G. Pourceau, A. Meyer, J. J. Vasseur and F. Morvan, J. Org. Chem., 2008, 73, 6014–6017 CrossRef CAS.
- F. Morvan, A. Meyer, A. Jochum, C. Sabin, Y. Chevolot, A. Imberty, J. P. Praly, J. J. Vasseur, E. Souteyrand and S. Vidal, Bioconjugate Chem., 2007, 18, 1637–1643 CrossRef CAS.
- G. Pourceau, A. Meyer, J. J. Vasseur and F. Morvan, J. Org. Chem., 2009, 74, 1218–1222 CrossRef CAS.
- R. T. Pon and S. Y. Yu, Nucleic Acids Res., 1997, 25, 3629–3635 CrossRef CAS.
- G. Pourceau, A. Meyer, Y. Chevolot, E. Souteyrand, J. J. Vasseur and F. Morvan, Bioconjugate Chem., 2010, 21, 1520–1529 CrossRef CAS.
- P. J. Garegg, T. Regberg, J. Stawinski and R. Stromberg, Chemica Scripta, 1985, 25, 280–282 CAS.
- B. C. Froehler, P. G. Ng and M. D. Matteucci, Nucleic Acids Res., 1986, 14, 5399–5407 CrossRef CAS.
- C. B. Reese and Q. L. Song, J. Chem. Soc., Perkin Trans. 1, 1999, 1477–1486 RSC.
- D. S. Sergueev and B. R. Shaw, J. Am. Chem. Soc., 1998, 120, 9417–9427 CrossRef CAS.
- B. Froehler, P. Ng and M. Matteucci, Nucleic Acids Res., 1988, 16, 4831–4839 CrossRef CAS.
- A. Laurent, M. Naval, F. Debart, J. J. Vasseur and B. Rayner, Nucleic Acids Res., 1999, 27, 4151–4159 CrossRef CAS.
- T. Michel, C. Martinand-Mari, F. Debart, B. Lebleu, I. Robbins and J. J. Vasseur, Nucleic Acids Res., 2003, 31, 5282–5290 CrossRef CAS.
- Y. Chevolot, J. Zhang, A. Meyer, A. Goudot, S. Rouanet, S. Vidal, G. Pourceau, J. P. Cloarec, J. P. Praly, E. Souteyrand, J. J. Vasseur and F. Morvan, Chem. Commun., 2011, 47, 8826–8828 RSC.
- C. Bouillon, A. Meyer, S. Vidal, A. Jochum, Y. Chevolot, J. P. Cloarec, J. P. Praly, J. J. Vasseur and F. Morvan, J. Org. Chem., 2006, 71, 4700–4702 CrossRef CAS.
- Y. Chevolot, C. Bouillon, S. Vidal, F. Morvan, A. Meyer, J. P. Cloarec, A. Jochum, J. P. Praly, J. J. Vasseur and E. Souteyrand, Angew. Chem., Int. Ed., 2007, 46, 2398–2402 CrossRef CAS.
- A. Meyer, F. Morvan and J. J. Vasseur, Tetrahedron Lett., 2004, 45, 3745–3748 CrossRef CAS.
- F. Ferreira, A. Meyer, J. J. Vasseur and F. Morvan, J. Org. Chem., 2005, 70, 9198–9206 CrossRef CAS.
- A. Meyer, C. Bouillon, S. Vidal, J. J. Vasseur and F. Morvan, Tetrahedron Lett., 2006, 47, 8867–8871 CrossRef CAS.
- A. Meyer, G. Pourceau, J. J. Vasseur and F. Morvan, J. Org. Chem., 2010, 75, 6689–6692 CrossRef CAS.
- L. Moni, G. Pourceau, J. Zhang, A. Meyer, S. Vidal, E. Souteyrand, A. Dondoni, F. Morvan, Y. Chevolot, J. J. Vasseur and A. Marra, ChemBioChem, 2009, 10, 1369–1378 CrossRef CAS.
- G. Mulvey, P. I. Kitov, P. Marcato, D. R. Bundle and G. D. Armstrong, Biochimie, 2001, 83, 841–847 CrossRef CAS.
- A. Bernardi, D. Arosio, D. Potenza, I. Sanchez-Medina, S. Mari, F. J. Canada and J. Jimenez-Barbero, Chem.–Eur. J., 2004, 10, 4395–4406 CrossRef CAS.
- M. Galibert, P. Dumy and D. Boturyn, Angew. Chem., Int. Ed., 2009, 48, 2576–2579 CrossRef CAS.
- A. Meyer, N. Spinelli, P. Dumy, J. J. Vasseur, F. Morvan and E. Defrancq, J. Org. Chem., 2010, 75, 3927–3930 CrossRef CAS.
- M. Karskela, M. Helkearo, P. Virta and H. Lonnberg, Bioconjugate Chem., 2010, 21, 748–755 CrossRef CAS.
- C. Kieburg, M. Dubber and T. K. Lindhorst, Synlett, 1997, 1447–1449 CAS.
- M. Dubber and T. K. Lindhorst, Carbohydr. Res., 1998, 310, 35–41 CrossRef CAS.
- M. Dubber and T. K. Lindhorst, Chem. Commun., 1998, 1265–1266 RSC.
- T. K. Lindhorst, in Host–Guest Chemistry, ed. S. Penades, Springer-Verlag, Berlin Heidelberg New York, 2002, vol. 218, pp. 201-235 Search PubMed.
- M. Kohn, J. M. Benito, C. O. Mellet, T. K. Lindhorst and J. M. G. Fernandez, ChemBioChem, 2004, 5, 771–777 CrossRef.
- B. Gerland, A. Goudot, G. Pourceau, A. Meyer, S. Vidal, E. Souteyrand, J. J. Vasseur, Y. Chevolot and F. Morvan, J. Org. Chem., 2012, 77, 7620–7626 CrossRef CAS.
- J. C. Bologna, F. Morvan and J. L. Imbach, Eur. J. Org. Chem., 1999, 2353–2358 CrossRef CAS.
- O. Sulak, E. Lameignere, M. Wimmerova and A. Imberty, in Carbohydrate Chemistry, ed. A. P. Rauter and L. Lindhorst, 2009, pp. 356-371 Search PubMed.
- T. Horlacher, M. A. Oberli, D. B. Werz, L. Krock, S. Bufali, R. Mishra, J. Sobek, K. Simons, M. Hirashima, T. Niki and P. H. Seeberger, ChemBioChem, 2010, 11, 1563–1573 CrossRef CAS.
- D. Leonard, Y. Chevolot, O. Bucher, W. Haenni, H. Sigrist and H. J. Mathieu, Surf. Interface Anal., 1998, 26, 793–799 CrossRef CAS.
- D. Léonard, Y. Chevolot, O. Bucher, H. Sigrist and H.-J. Mathieu, Surf. Interface Anal., 1998, 26, 783–792 CrossRef.
- Y. Chevolot, O. Bucher, D. Léonard, H.-J. Mathieu and H. Sigrist, Bioconjugate Chem., 1999, 10, 169–175 CrossRef CAS.
- Y. Chevolot, J. Martins, N. Milosevic, D. Leonard, S. Zeng, M. Malissard, E. Berger, G. P. Maier, H. Mathieu, J. D. Crout and H. H. Sigrist, Bioorg. Med. Chem., 2001, 9, 2943–2953 CrossRef CAS.
- D. Léonard, Y. Chevolot, F. Heger, J. Martins, D. Crout, H. G. H. Sigrist and H. Mathieu, Surf. Interface Anal., 2001, 31, 457–464 CrossRef.
- F. Fazio, M. Bryan, C. O. Blixt, J. Paulson and C.-H. Wong, J. Am. Chem. Soc., 2002, 124, 14397–14402 CrossRef CAS.
- S. Fukui, T. Feizi, C. Galustian, A. M. Lawson and W. Chai, Nat. Biotechnol., 2002, 20, 1011–1017 CrossRef CAS.
- B. Houseman and T. M. Mrksich, Chem. Biol., 2002, 9, 443–454 CrossRef CAS.
- D. Wang, S. Liu, B. J. Trummer, C. Deng and A. Wang, Nat. Biotechnol., 2002, 20, 275–281 CrossRef CAS.
- T. Feizi, F. Fazio, W. Chai and C.-H. Wong, Curr. Opin. Struct. Biol., 2003, 13, 637–645 CrossRef CAS.
- E. A. Smith, W. D. Thomas, L. L. Kiessling and R. M. Corn, J. Am. Chem. Soc., 2003, 125, 6140–6148 CrossRef CAS.
- D. Wang, Proteomics, 2003, 3, 2167–2175 CrossRef CAS.
- C. Galustian, C. G. Park, W. Chai, M. Kiso, S. A. Bruening, Y. S. Kang, R. M. Steinman and T. Feizi, Int. Immunol., 2004, 16, 853–866 CrossRef CAS.
- S. Park, M. R. Lee, S. J. Pyo and I. Shin, J. Am. Chem. Soc., 2004, 126, 4812–4819 CrossRef CAS.
- D. Ratner, M. E. Adams, W. J. Su, B. O'Keefe, R. M. Mrksich and P. Seeberger, ChemBioChem, 2004, 5, 379–383 CrossRef CAS.
- O. Blixt, S. Head, T. Mondala, C. Scanlan, M. Huflejt, E. R. Alvarez, M. Bryan, C. F. Fazio, D. Calarese, J. Stevens, N. Razi, D. J. Stevens, J. J. Skehel, I. van Die, D. Burton, R. I. Wilson, A. R. Cummings, N. Bovin, C.-H. Wong and J. C. Paulson, Proc. Natl. Acad. Sci. U. S. A., 2004, 10, 17033–17038 CrossRef.
- D. B. Werz and P. H. Seeberger, Chem.–Eur. J., 2005, 11, 3194–3206 CrossRef CAS.
- M. S. Timmer, B. L. Stocker and P. H. Seeberger, Curr. Opin. Chem. Biol., 2007, 11, 59–65 CrossRef CAS.
- O. Oyelaran and J. C. Gildersleeve, Expert Rev. Vaccines, 2007, 6, 957–969 CrossRef CAS.
- Y. Zhang, Q. Li, L. G. Rodriguez and J. C. Gildersleeve, J. Am. Chem. Soc., 2010, 132, 9653–9662 CrossRef CAS.
- A. Gordus and G. MacBeath, J. Am. Chem. Soc., 2006, 128, 13668–13669 CrossRef CAS.
- X. Song, Y. Lasanajak, B. Xia, D. F. Smith and R. D. Cummings, ACS Chem. Biol., 2009, 4, 741–750 CrossRef CAS.
- X. Song, B. Xia, S. R. Stowell, Y. Lasanajak, D. F. Smith and R. D. Cummings, Chem. Biol., 2009, 16, 36–47 CrossRef CAS.
- D. J. Scurr, T. Horlacher, M. A. Oberli, D. B. Werz, L. Kroeck, S. Bufali, P. H. Seeberger, A. G. Shard and M. R. Alexander, Langmuir, 2010, 26, 17143–17155 CrossRef CAS.
- A. Palma, S. T. Feizi, Y. Zhang, M. Stoll, S. A. Lawson, M. E. Diaz-Rodrıguez, M. Campanero-Rhodes, A. J. Costa, S. Gordon, G. Brown and D. C. Wengang, J. Biol. Chem., 2006, 281, 5771–5779 CrossRef CAS.
- F. Jaipuri, A. B. Collet, Y. M. Collet and N. L. Pohl, Angew. Chem., Int. Ed., 2008, 47, 1707–1710 CrossRef CAS.
- K. Larsen, M. Thygesen, B. F. Guillaumie, W. G. T. Willats and K. J. Jensen, Carbohydr. Res., 2006, 341, 1209–1234 CrossRef CAS.
- M. Dhayal and D. M. Ratner, Langmuir, 2009, 25, 2181–2187 CrossRef CAS.
- J. Manimala, C. Z. Li, A. Jain, S. VedBrat and J. C. Gildersleeve, ChemBioChem, 2005 Search PubMed.
- J. Manimala, C. T. Roach, A. Z. Li and J. C. Gildersleeve, Angew. Chem., Int. Ed., 2006, 45, 3607–3610 CrossRef CAS.
- M. Schwarz, L. Spector, A. Gargir, A. Shtevi, M. Gortler, R. Altstock, T. A. Dukler and A.N. Dotan, Glycobiology, 2003, 13, 749–754 CrossRef CAS.
- M. Biskup, B. J. Müller, U. R. Weingart and R. R. Schmidt, ChemBioChem, 2005, 6, 1007–1015 CrossRef CAS.
- M. R. Lee and I. Shin, Angew. Chem., Int. Ed., 2005, 44, 2881–2884 CrossRef CAS.
- M. Kohn, R. Wacker, C. Peters, H. Schröder, L. Soulère, R. Breinbauer, C. Niemeyer and M. H. Waldmann, Angew. Chem., Int. Ed., 2003, 42, 5830–5834 CrossRef.
- M. Bryan, C. F. Fazio, H.-K. Lee, C.-Y. Huang, A. Chang, M. Best, D., D. Calarese, A. O. Blixt, J. Paulson, C. D. Burton, I. Wilson and A. C.-H. Wong, J. Am. Chem. Soc., 2004, 126, 8640–8641 CrossRef CAS.
- J. Seibel, H. Hellmuth, B. Hofer, A.-M. Kicinska and B. Schmalbruch, ChemBioChem, 2006, 7, 310–320 CrossRef CAS.
- C. Y. Huang, D. A. Thayer, A. Y. Chang, M. D. Best, J. Hoffmann, S. Head and C. H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15–20 CrossRef CAS.
- B. Bochner, S. R. Alvarez, A. P. Mehta, N. Bovin, V. O. Blixt, J. White and R. R. Schnaar, J. Biol. Chem., 2005, 280, 4307–4312 CrossRef CAS.
- E. Muñoz, M. H. Yu, J. Hallock, R. Edens and E. R. Linhardt, Anal. Biochem., 2005, 343, 176–178 CrossRef.
- M. J. Linman, J. D. Taylor, H. Yu, X. Chen and Q. Cheng, Anal. Chem., 2008, 80, 4007–4013 CrossRef CAS.
- Y. Wang and T. L. Sheppard, Bioconjugate Chem., 2003, 14, 1314–1322 CrossRef CAS.
- C. M. Niemeyer, Angew. Chem., Int. Ed., 2010, 49, 2–19 CrossRef.
- J. Harris, D. E. Mason, J. Li, K. W. Burdick, B. J. Backes, T. Chen, A. Shipway, G. Van Heeke, L. Gough, A. Ghaemmaghami, F. Shakib, F. Debaene and N. Winssinger, Chem. Biol., 2004, 11, 1361–1372 CrossRef CAS.
- F. Debaene and N. Winssinger, Methods Mol. Biol., 2009, 570, 299–307 CAS.
- C. F. W. Becker, R. Wacker, W. Bouschen, R. Seidel, B. Kolaric, P. Lang, H. Schroeder, O. Müller, C. M. Niemeyer, B. Spengler, R. S. Goody and M. Engelhard, Angew. Chem., Int. Ed., 2005, 44, 7635–7639 CrossRef CAS.
- J. L. Wang, D. K. Xu and H. Y. Chen, Electrochem. Commun., 2009, 11, 1627–1630 CrossRef CAS.
- R. Wacker and C. M. Niemeyer, ChemBioChem, 2004, 5, 453–459 CrossRef CAS.
- H. Schroeder, B. Ellinger, C. F. W. Becker, H. Waldmann and C. M. Niemeyer, Angew. Chem., Int. Ed., 2007, 46, 4180–4183 CrossRef CAS.
- J. Zhang, G. Pourceau, A. Meyer, S. Vidal, J. P. Praly, E. Souteyrand, J. J. Vasseur, F. Morvan and Y. Chevolot, Biosens. Bioelectron., 2009, 24, 2515–2521 CrossRef CAS.
- S. Park and I. Shin, Org. Lett., 2007, 9, 1675–1678 CrossRef CAS.
- P.-H. Liang, S.-K. Wang and C.-H. Wong, J. Am. Chem. Soc., 2007, 129, 11177–11184 CrossRef CAS.
- J. Zhang, G. Pourceau, A. Meyer, S. Vidal, J. P. Praly, E. Souteyrand, J. J. Vasseur, F. Morvan and Y. Chevolot, Chem. Commun., 2009, 6795–6797 RSC.
- A. Goudot, G. Pourceau, A. Meyer, T. Gehin, S. Vidal, J. J. Vasseur, F. Morvan, E. Souteyrand and Y. Chevolot, Biosens. Bioelectron., 2012 DOI:10.1016/j.bios.2012.07.003.
- R. Mazurczyk, G. E. Khoury, V. Dugas, B. Hannes, E. Laurenceau, M. Cabrera, S. Krawczyk, E. Souteyrand, J. P. Cloarec and Y. Chevolot, Sens. Actuators, B, 2008, 128, 552–559 CrossRef.
- J. Vieillard, R. Mazurczyk, L. L. Boum, A. Bouchard, Y. Chevolot, P. Cremillieu, B. Hannes and S. Krawczyk, Microelectron. Eng., 2008, 85, 465–469 CrossRef CAS.
- N. R. Markham and M. Zuker, Nucleic Acids Res., 2005, 33, W577–W581 CrossRef CAS.
- O. V. Matveeva, S. A. Shabalina, V. A. Nemtsov, A. D. Tsodikov, R. F. Gesteland and J. F. Atkins, Nucleic Acids Res., 2003, 31, 4211–4217 CrossRef CAS.
- N. Sphyris, J. M. Lord, R. Wales and L. M. Roberts, J. Biol. Chem., 1995, 270, 20292–20297 CrossRef CAS.
- R. Hegde and S. K. Podder, Eur. J. Biochem., 1998, 254, 596–601 CAS.
- S. Sharma, S. Bharadwaj, A. Surolia and S. K. Podder, Biochem. J., 1998, 333, 539–542 CAS.
- B. Ernst and J. L. Magnani, Nat. Rev. Drug Discovery, 2009, 8, 661–677 CrossRef CAS.
- D. Deniaud, K. Julienne and S. G. Gouin, Org. Biomol. Chem., 2011, 9, 966–979 CAS.
- S. Cecioni, R. Lalor, B. Blanchard, J. P. Praly, A. Imberty, S. E. Matthews and S. Vidal, Chem.–Eur. J., 2009, 15, 13232–13240 CrossRef CAS.
- B. Gerland, A. Goudot, G. Pourceau, A. Meyer, V. Dugas, S. Cecioni, S. Vidal, E. Souteyrand, J. J. Vasseur, Y. Chevolot and F. Morvan, Bioconjugate Chem., 2012, 23, 1534–1547 CrossRef CAS.
Footnote |
| † Dedicated to the memory of Professor H. Gobind Khorana. |
| This journal is © The Royal Society of Chemistry 2012 |