Synthesis of poly(ethyl acrylate-co-allyl acrylates) from acrylate mixtures prepared by a continuous solvent-free enzymatic process†
Edinson
Yara-Varón
a,
Jordi
Eras Joli
a,
Mercè
Balcells
a,
Mercè
Torres
b and
Ramon
Canela-Garayoa
*a
aDepartment of Chemistry, Lleida University, ETSEA, Av. Rovira Roure 191, 25198, Lleida, Spain. E-mail: canela@quimica.udl.cat; Fax: +34 973238264; Tel: +34 973702843.
bDepartment of Food Science and Technology, Lleida University, ETSEA, Av. Rovira Roure 191, 25198, Lleida, Spain
First published on 25th July 2012
Abstract
A miniaturized, packed-bed, continuous-flow reactor to carry out the enzymatic interesterification of ethyl acrylate with diverse allyl aliphatic esters is described. CALB (lipase B from Candida antarctica immobilized on a macroporous acrylic resin) was used as a biocatalyst in a solvent-free system. Allyl acrylate was produced with a conversion of 45% and productivity of 48.3 mg (g biocatalyst)−1 min−1. Then, polymeric materials were prepared from mixtures of acrylate esters. Copolymerization was achieved both in bulk and using water as the solvent. Depending on the polymerizing system used, polymerization yields were 83% and 70% after 2 h and 18 h, respectively. Successful synthesis in a miniaturized continuous-flow reactor allows the use of allyl esters obtained from renewable sources as starting materials.
Introduction
Increased petroleum prices and the probability that continued use of petroleum-based feedstocks will lead to an unacceptable amount of greenhouse gas emissions have renewed interest in the use of biomass as a primary source for the chemical industry. Thus, despite the pioneering studies of Stearn et al.1 in 1940 on the synthesis of methyl acrylate from lactic acid, only recently has this natural compound aroused interest as a starting material for acrylic acid production.2–4 Acrylics are employed to produce dyes, paper, textiles, glues, adhesives, binders, paints, dispersants, thickeners, and flocculants.5,6 More than half of the world production of acrylic acid is used to synthesize various acrylic acid esters, mainly methyl, ethyl, n-butyl, 2-ethylhexyl, and allyl acrylates.5 Allyl acrylate is used for preparing copolymeric materials.6–11 It is also common in dentistry.5Acrylates are highly sensitive to the conditions used in traditional chemical esterification. This process occurs in the presence of both alkaline and acidic catalysts.12–14 However, the traditional reaction requires the use of special operating procedures and harsh catalysts.15 Although these chemical processes are used on an industrial scale, the reaction has several drawbacks: it is energy intensive, the acidic or alkaline catalyst has to be removed from the product, and alkaline waste water requires treatment.16 To overcome these problems, the use of lipases for the acrylate transacylation has been proposed.15,17–20 Industrial interest in enzymatic acrylation is also reflected in two recent patents related to the subject.21,22
We recently described the synthesis of allyl acrylate starting from ethyl acrylate and allyl fatty esters (Scheme 1). These esters in turn can be prepared in two steps using crude glycerol from the biodiesel industry and vegetable oils or fat, some of them obtained from agri-food waste.23
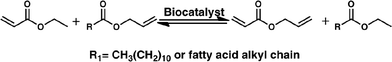 | ||
| Scheme 1 Allyl acrylate synthesis from allyl dodecanoate and ethyl acrylate using a biocatalyst in a solvent-free system. | ||
Recently, we described a solvent-free interesterification reaction between ethyl acrylate and allyl esters in a batch reactor using diverse biocatalysts. Of these, CALB (lipase B from Candida antarctica immobilized on a macroporous acrylic resin) showed the best performance.24
Here, we sought to study the enzymatic interesterification of ethyl acrylate with diverse allyl aliphatic esters using CALB in a miniaturized, packed-bed, continuous-flow reactor. The process was carried out without solvent, as in the batch reactor. Carrying out the reaction in a solvent-free system may help to develop a process with a low E-factor.25 The flow reactor should allow the synthesis of sufficient allyl acrylate to prepare acrylic copolymers.
The main advantages of flow systems are facile automation, reproducibility, and process reliability, because they allow the maintenance of constant reaction parameters (e.g., temperature, time, amount of reagents, solvent).26–29 Among these systems, the miniaturized continuous-flow reactor shows promise as a novel method to achieve these aims. Recently, Woodcock et al.30 described the synthesis of a series of alkyl esters using a biocatalyst in a packed-bed, miniaturized, continuous-flow reactor. In fact, many groups have begun the task of transferring organic synthesis methodologies from batch to miniaturized flow reactors.31 The introduction of ‘flow chemistry’ into organic synthesis laboratories has begun to change the way in which chemical reactions are performed.32–37
Experimental
Materials
Dodecanoic acid, ethyl dodecanoate, allyl methacrylate, ethyl methacrylate, CALB, benzoyl peroxide (BPO) 75%, ascorbic acid (99% purity), and poly(vinyl pyrrolidone) were purchased from Sigma-Aldrich (Sigma-Aldrich Quimica, S.A., Madrid, Spain). Ethyl acrylate and butyl acrylate were from Fluka (Sigma-Aldrich, Madrid, Spain). Allyl acrylate was supplied by Alfa Aesar (Barcelona, Spain), and hexane was purchased from J.T.Baker (Quimega, Lleida, Spain).The biocatalyst is produced through the physical adsorption of free CALB onto polyacrylate beads (10% w/w lipase B physically adsorbed within 90% w/w Lewatit VPOC 1600 support), which consist of poly(methyl methacrylate) cross-linked with divinylbenzene. The spherical beads have an average particle size of 0.315–1.0 mm (>80%), bulk density of 0.65–0.8 g cm−3, high surface area of ∼130 m2 g−1 (BET), and high thermal stability (−20 to 100 °C).16,38
Procedure for the preparation of allyl esters
Allyl esters were prepared from glycerol and either dodecanoic acid, soybean oil, or waste vegetable frying oils, as described previously.39,40 Crude compounds were purified by distillation under vacuum with a Büchi Kugelrohr apparatus (Massó Analítica S.A., Barcelona, Spain).Continuous-flow system reactions
The mini packed-bed reactor consisted of a glass column (inner diameter: 3 mm; total length: 100 mm; packed length: 90 mm; inner volume: 0.70 mL) filled with 240 mg of CALB supported on acrylic resin. The glass column was packed manually with the immobilized enzyme, which was then fixed in place using plugs of glass wool (Fig. 1). A mixture of ethyl acrylate (3.00 g, 30 mmol) and allyl fatty esters was pumped through the column reactor at a constant flow rate (FRX System, Syrris Ltd, Royston, UK).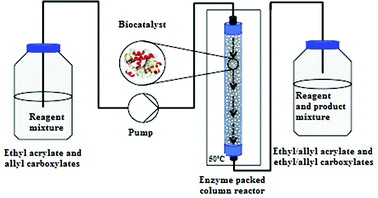 | ||
| Fig. 1 Scheme of the continuous-flow system applied. | ||
The column reactor was heated with an FRX Volcano Column Adaptor (Syrris) and a digitally controlled RCT basic hotplate (IKA-Werke GmbH & Co., KG, Staufen, Germany) with external Pt 100 sensor for optimum control of temperature. A digital control with an external sensor ensured that the temperature setting was correctly maintained in the column. The reaction was performed in a solvent-free system.
Scope of the reaction
Using a mixture of allyl dodecanoate and ethyl acrylate, we studied the following parameters: a) mole ratio of reactants (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5; 1:0.75; 1:1; 1:2; 1:3; 1:5, and 1:10), temperature (50 and 60 °C), amount of biocatalyst (240 mg and 480 mg) and flow rate (10, 40, and 80 μL min−1). The viscosity of the mixtures at 50 °C was an average of 1.4 × 10−6 m2 s−1, as measured by a thermostated Ubbelohde viscometer Comecta (JP Selecta, Abrera, Barcelona, Spain). The water activity (aw) of CALB and the reagents in all of the experiments was set by equilibration with aqueous saturated solutions of MgCl2. The aw was measured using an Aqua Lab series 3TE (Decagon Devices Inc., Pullman, WA, USA).24 Once the optimal parameters had been fixed, we examined the reaction using allyl esters prepared from soybean oil and from waste frying vegetable oil. The reverse reaction between ethyl dodecanoate and allyl acrylate was also studied. Finally, ethyl methacrylate was used instead of ethyl acrylate. All of the reactions were carried out in triplicate.
:0.5; 1:0.75; 1:1; 1:2; 1:3; 1:5, and 1:10), temperature (50 and 60 °C), amount of biocatalyst (240 mg and 480 mg) and flow rate (10, 40, and 80 μL min−1). The viscosity of the mixtures at 50 °C was an average of 1.4 × 10−6 m2 s−1, as measured by a thermostated Ubbelohde viscometer Comecta (JP Selecta, Abrera, Barcelona, Spain). The water activity (aw) of CALB and the reagents in all of the experiments was set by equilibration with aqueous saturated solutions of MgCl2. The aw was measured using an Aqua Lab series 3TE (Decagon Devices Inc., Pullman, WA, USA).24 Once the optimal parameters had been fixed, we examined the reaction using allyl esters prepared from soybean oil and from waste frying vegetable oil. The reverse reaction between ethyl dodecanoate and allyl acrylate was also studied. Finally, ethyl methacrylate was used instead of ethyl acrylate. All of the reactions were carried out in triplicate.
Monitoring CALB stability at reaction conditions
To evaluate the stability of the biocatalyst, the reaction was carried out in the continuous-flow system with ethyl acrylate and allyl dodecanoate at 50 °C for 144 h. Samples were collected at the column reactor outlet at a range of time intervals and analysed as indicated below. All of the reactions were performed in triplicate.Recovery of acrylate esters
A mixture of ethyl acrylate (9.00 g, 90 mmol) and allyl dodecanoate (21.63 g, 90 mmol) was pumped through the column reactor (3 mm diameter × 100 mm length) under a constant flow rate of 10 μL min−1 at 50 °C. The recovered liquid mixture was distilled under vacuum at 29 °C and 18 Torr. We obtained 8.23 g of a mixture of allyl acrylate and ethyl acrylate (4:7 ratio). This amount corresponds to a molar recovery of 87.5% with respect to the initial amount of ethyl acrylate (31.8% yield of allyl acrylate).
Preparation of a polyacrylate
Solubility tests
The solubility of the four copolymers prepared was studied using a set of solvents of various polarities (hexane, toluene, benzene, ethyl ether, tert-butyl methyl ether, dioxane, tetrahydrofuran, ethyl acetate, chloroform, dichloromethane, propanone, dimethyl sulfoxide, acetic anhydride, acetic acid, ethanol, methanol, and water).Analysis
The progress of each reaction was determined by gas chromatography (GC) using an Agilent (Barcelona, Spain) HP6890 series GC coupled to a flame ionization detector (FID). The analytical column was a 30 m × 0.25 mm fused silica capillary coated with a 0.20 μm film of poly(80% biscyanopropyl–20% cyanopropylphenyl siloxane) (SP-2330; Supelco, Madrid, Spain). The temperature program used was 40 °C for 5 min; then the temperature was increased at a rate of 20 °C min−1 until holding at the final temperature of 225 °C for 3 min. A 1:20 split injection ratio was used. Hydrogen was used as the carrier gas at a constant pressure of 620 kPa. The injection volume was 1 μL. The injection system was held at 250 °C and the FID system at 280 °C. Quantification was performed by a conventional external standard method using the corresponding acrylic ester standards.
Solids were vacuum-dried and analysed using a Magna IR 560 Nicolet FT-IR spectrometer, an alpha-300R Witec confocal Raman spectrometer and a Varian 400 NMR spectrometer. The C:H ratio of each solid was determined by combustion elemental analysis in a Carlo Erba Instruments EA 1108.42
Results and discussion
Condition optimization
Interesterification studies were started using a 1:1 mole ratio of ethyl acrylate:allyl dodecanoate, 240 mg of biocatalyst, and the minimum flow rate allowed for our system (10 μL min−1).
Column temperatures of 50 and 60 °C were studied. Although a higher conversion was obtained at 60 °C, the highest yield (39.8%) was obtained at 50 °C (Table 1). This inconsistency between conversion and yield could be explained by noting that this biocatalyst is an enzyme adsorbed on a macroporous acrylic resin. Hence, a portion of the acrylic compounds could be adsorbed on the resin beads, resulting in an increase in conversion results (yield of ethyl acrylate recovered) and a decrease in allyl acrylate yield. Indeed, this decrease could be due to a loss of enzyme activity when the temperature rises43,44 or an increase of allyl acrylate retention by the support,45–47 or both. Consequently, all subsequent experiments were conducted at this temperature. The yield obtained was very close to that achieved using a batch system with the same reaction conditions: 0.290 mL of reactant mixture, 240 mg of biocatalyst, temperature of 50 °C and reaction time of 29 min. This reaction time is equivalent to the 29 min of residence time in continuous flow calculated with the ε/q expression (ε is the catalyst bed porosity, determined experimentally, and q is the flow rate).48,49
:1 mole ratio of ethyl acrylate:allyl dodecanoate, CALB, no solvent
| Biocatalyst (mg) | T/°C | Flow rate (μL min−1) | Conversiona (%) | Allyl acrylate yielda (%) |
|---|---|---|---|---|
| a Conversion and yield calculated using a GC–FID system. b 40 mmol of ethyl acrylate. c Two 3 × 100 mm column reactor cascade. d Reaction in a batch system and reaction time 30 min. | ||||
| 240 | 50 | 10 | 45.9 ± 1.3 | 39.8 ± 0.6 |
| 240 | 60 | 10 | 48.7 ± 0.7 | 32.2 ± 0.6 |
| 240 | 50 | 40 | 12.6 ± 1.5 | 10.4 ± 0.8 |
| 240 | 50 | 80 | 13.8 ± 0.8 | 12.3 ± 0.9 |
| 480bc | 50 | 10 | 51.8 ± 0.5 | 21.8 ± 0.3 |
| 240d | 50 | — | 43.7 ± 1.4 | 39.2 ± 1.7 |
In new sets of experiments, we increased the amount of the biocatalyst using a two-column reactor cascade, which increased the residence time of the system. In this case, 40 mmol of each reagent were used. Nevertheless, although conversion increased to around 52% (Table 1), yields dropped to 21.8%. These results indicate that a high proportion of the chemicals was lost during these processes. This loss was possibly a result of the higher ratio of the biocatalyst and its supporting polymeric material in relation to the reaction mixture. As mentioned above, a portion of the acrylic compounds could be adsorbed on the resin beads, producing an increase in the conversion rate and a decrease in the yield. Given that an increase of only 6% (from 46% to 52%) does not justify the use of a double amount of biocatalyst, we performed the following experiments using a single 3 mm × 100 mm column reactor and 240 mg of biocatalyst. The conversion (45.9%) and yield (39.8%) for a flow rate of 10 μL min−1 were considerably higher than those achieved for flow rates of 40 and 80 μL min−1 (12.6 and 13.8% conversions and 10.4 and 12.3% yields, respectively) (Table 1). Although higher flow rates can diminish axial dispersion phenomena and interaction with a stagnant zone in a packed-bed reactor, they also reduce the residence time of the reagents in the column reactor.50 While the decrease of axial dispersion should improve conversions, the reduction in the residence time should lead to a decrease in this parameter. Given the result obtained in these experiments, the increase in residence time had a higher effect on the percentage conversion than the expected decrease in the axial dispersion. The decrease in conversion by increasing the flow rate has been extensively reported in continuous enzyme-catalysed reactions in small fixed-bed reactors. Xi and Xu51 described a large drop in the conversion rates in their study of the preparation of enantiopure (S)-ketoprofen by immobilized Candida rugosa lipase in a packed-bed reactor. The conversion decreased from 40% (98 L min−1) to about 10% at 650 or 800 L min−1.
Although in our batch studies we found that the 1:1 mole ratio gave better results than the 1:5 mole ratio, we performed a set of experiments using various ethyl acrylate:allyl dodecanoate mole ratios. The interesterification reactions were carried out using 240 mg of biocatalyst (3 mm × 100 mm column) at a flow rate of 10 μL min−1 at 50 °C. As in the batch reaction, the 1:1 mole ratio showed the best performance (Fig. 2). Mole ratios of 1:2 or 2:1 gave similar results with respect to allyl acrylate yields. While the increase in the allyl dodecanoate ratio from 1:0.5 to 1:0.75 resulted in an enhanced allyl acrylate yield, subsequent increases from 1:3 to 1:10 caused a clear drop in these yields. These results suggest that the 45.9% conversion obtained with the 1:1 mole ratio is a conversion near the equilibrium between the two acrylates present in this interesterification.
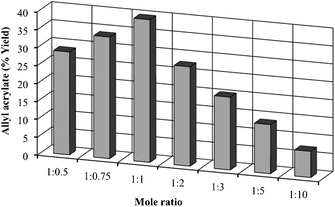 | ||
| Fig. 2 Effect of the mole ratio (ethyl acrylate:allyl dodecanoate) on the synthesis of allyl acrylate under the fixed conditions of temperature (50 °C), lipase CALB (240 mg), and flow rate (10 μL min−1). 30 mmol of ethyl acrylate were used in each experiment. | ||
To confirm this hypothesis, we performed the inverse reaction starting with a 1:1 mole ratio of allyl acrylate and ethyl dodecanoate. A final percentage of 45.7 ± 1.5% for the allyl acrylate was achieved, thereby confirming the hypothesis. Nevertheless, this degree of interesterification at 1:1 ratios of both starting esters could also be attributed to competition between both esters for the catalytic centre. Hence, an increase in the mole fraction of one of the starting esters should produce a decrease in the ability of the other starting ester to reach the corresponding catalytic centre. In consequence, the interesterification yield would decrease.
Reaction scope
Table 2 shows the conversion and yields obtained when other reagents were used. The best mole ratio, amount of enzyme, column reactor size, temperature, and flow rate found previously were used in subsequent experiments. Although lower conversions were observed using allyl esters of fatty acids with longer alkyl chains (37.2% for allyl esters obtained from soybean oil and 36% for those obtained from waste frying vegetable oil), the allyl acrylate yields (around 37%) were very similar, in spite of the use of allyl fatty esters as reagents. This finding indicates that the longer fatty acids are more suitable solvents for the acrylate esters present in the medium than are dodecanoate esters. However, interesterification using ethyl methacrylate instead of ethyl acrylate gave a 27.6 and 25.3% lower conversion and yield, respectively. In this case, the reverse reaction (allyl methacrylate and ethyl dodecanoate as starting reagents) resulted in 55% of the allyl methacrylate remaining. This result indicates that equilibrium in the direct reaction was not reached.| Allyl esters | Ethyl esters | Allylic product | Conversion (%) | Final allyl acrylates (% yield) |
|---|---|---|---|---|
|
a Reaction conditions: continuous-flow rate at 10 μL min−1, 240 mg of supported lipase CALB, 50 °C, no solvent, 1:1 mole ratio allyl esters:ethyl esters (allyl dodecanoate (1a), allyl esters from soybean (1b), allyl esters from waste vegetable oil (1c), allyl acrylate (1d), allyl methacrylate (1e), ethyl acrylate (2a), ethyl dodecanoate (2b) and ethyl methacrylate (2c)).
b Remaining allyl acrylate.
c Remaining allyl methacrylate.
|
||||
| 1a | 2a | Allyl acrylate | 45.9 ± 1.3 | 39.8 ± 0.6 |
| 1b | 2a | Allyl acrylate | 38.7 ± 1.4 | 37.1 ± 1.3 |
| 1c | 2a | Allyl acrylate | 38.8 ± 1.5 | 37.6 ± 0.9 |
| 1d | 2b | Allyl dodecanoate | 45.7 ± 1.4 | 45.7 ± 1.5b |
| 1a | 2c | Allyl methacrylate | 27.6 ± 1.6 | 25.3 ± 1.6 |
| 1e | 2b | Allyl dodecanoate | 55.3 ± 1.4 | 49.5 ± 1.7c |
Stability of the biocatalyst over an extended period of operation
Although the interesterification reaction was still present after 144 h, the amount of ester produced during every 24 h period was lower than in the previous period (Fig. 3), showing a continuous decrease in the biocatalytic activity of the supported enzyme. Nevertheless, the conversion obtained during the initial 24 h of reaction was relatively constant, showing a maximum after 18 h of reaction. This length of time was necessary to pass 60 mmol of reagent mixture through the column reactor using a 1:1 mole ratio.
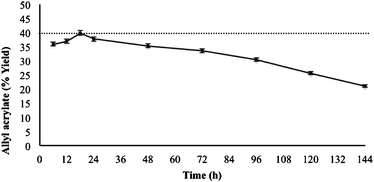 | ||
| Fig. 3 Effect of time period on the synthesis of allyl acrylate under the fixed conditions of temperature (50 °C), lipase CALB (240 mg), and flow rate (10 μL min−1). 40 mmol of ethyl acrylate passed through the flow reactor every 24 h. The maximum conversion is indicated with a dashed line. | ||
Progressive lipase inactivation in continuous enzyme-catalysed reactions in small fixed-bed reactors has been extensively reported. Loss of activity can be observed from a few hours to weeks after initial reaction. This gradual loss of activity is very common in lipase-catalysed reactions.45 Thus, Woodcock et al.,30 describing the synthesis of aliphatic esters using a small fixed-bed reactor, reported a loss of activity for some substrates after a 2 h reaction at a flow rate of 1 μL min−1. Biocatalytic activity was reported to decrease by 3% for the best substrates after 7.5 h of reaction. As far as we know, nobody has described the preparation of acrylic esters using a continuous-flow system.
To determine the potential for biocatalyst reuse after 144 h of reaction, it was washed several times with n-hexane and dried after this reaction period. The reactivated CALB was used in an identical experiment for 18 h of reaction time. The conversion was 43%, similar to the best conversion (46%) achieved using fresh enzyme. This result is very similar to those described by Osório et al.52
Preparation of a copolymer
Once the process was optimized and the scope of the reaction studied, it was pertinent to determine whether polymeric materials could be obtained from the mixture of acrylate esters prepared. For this purpose, we followed two approaches. First, the crude product of the reaction was distilled as described in the Experimental section and the recovered liquid was analysed by GC–FID, determining a 4:7 mole ratio of allyl acrylate:ethyl acrylate. This ratio was lower than that present in the crude product of the reaction (5:6). This distillate was used as the initial mixture for the polymerization. Second, the crude product of the reaction was also used without any further purification. The polymers were prepared using a typical radical catalysing polymerization method.11,53–56
Six experiments were carried out for bulk polymerization. Each initial mixture was shaken orbitally at 70 °C for 0.5, 1, and 2 h. The processes were quenched, and the white solids formed were washed until no monomer was observed by GC–FID in the washing solutions.
Fig. 4 shows the conversion and the yield of copolymer formed from two different starting materials (A: distilled mixture; B: crude reaction mixture). In both cases, the conversions increased with polymerization times. The two acrylic esters showed a polymerization yield of up to 80% after 2 h. Both ethyl acrylate and allyl acrylate were incorporated at a high proportion to the polymers formed, although the latter was incorporated in a higher ratio. When the crude reaction mixture was used, ethyl dodecanoate was almost fully recovered, whereas a small amount of allyl dodecanoate became part of the polymers prepared. These results are in accordance with the elemental analyses of the two copolymers. These analyses determined a 3:4 ratio between allyl acrylate and ethyl acrylate for the copolymer prepared from the mixture of the two acrylic esters. This ratio corresponds to an empirical formula of C38H59O14 (Elem. Anal. calculated for C38H59O14: C, 61.86; H, 7.75; O, 30.34. Found: C, 61.95; H, 7.71; O, 30.34%). In contrast, the elemental analysis of the copolymer prepared from the crude reaction mixture corresponds to an empirical formula of C31H48O10 (Elem. Anal. calculated for C31H48O10: C, 64.12; H, 8.33; O, 27.55. Found: C, 64.23; H, 8.44; O, 27.32%). From these results, we determined a 7:7:1 ratio for allyl acrylate:ethyl acrylate:allyl dodecanoate.
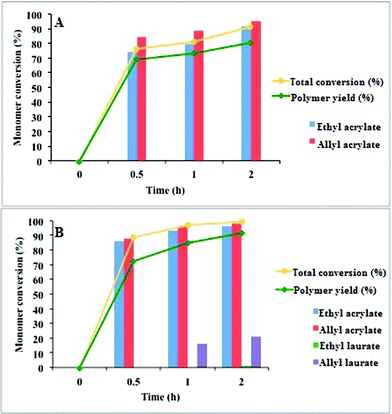 | ||
| Fig. 4 Influence of reaction time and starting material composition on the formation of the polymeric materials. The solid bars represent the percentage of monomer conversion. Solid lines show the progress of polymerization with time (conversion and copolymer yield based on the acrylate esters). | ||
Fig. 5 shows the FT-IR spectra of the two copolymers obtained after a 2 h reaction using the distilled fraction (A) and the crude reaction mixture (B). In both cases, characteristic IR bands corresponding to the presence of non-conjugated carboxylic esters (1734 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching; 1175–1160 and 1021 cm−1, C–O stretching), alkyl saturated chains (2970 cm−1, C–H stretching), and olefinic bonds (3088 cm−1, C–H stretching; and 1666–1620 cm−1, CC stretching) can be observed. The presence of these functional groups was also confirmed by the Raman spectra (1736 cm−1, CO stretching; 1170 and 972 cm−1, C–O stretching; 2982–2944 cm−1, C(sp3)–H stretching; 3100 cm−1, C(sp2)–H stretching; 1658–1620 cm−1, CC stretching), which in some cases (3100 cm−1 and 1658–1620 cm−1) showed stronger bands than those present in the FT-IR spectra.
O stretching; 1175–1160 and 1021 cm−1, C–O stretching), alkyl saturated chains (2970 cm−1, C–H stretching), and olefinic bonds (3088 cm−1, C–H stretching; and 1666–1620 cm−1, CC stretching) can be observed. The presence of these functional groups was also confirmed by the Raman spectra (1736 cm−1, CO stretching; 1170 and 972 cm−1, C–O stretching; 2982–2944 cm−1, C(sp3)–H stretching; 3100 cm−1, C(sp2)–H stretching; 1658–1620 cm−1, CC stretching), which in some cases (3100 cm−1 and 1658–1620 cm−1) showed stronger bands than those present in the FT-IR spectra.
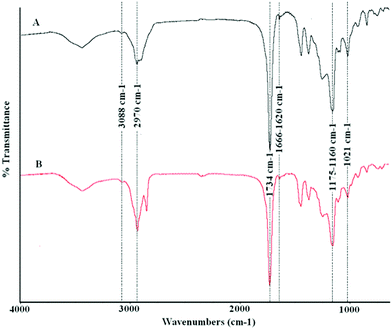 | ||
| Fig. 5 FT-IR spectra of the copolymers prepared from a distilled mixture of acrylic esters (A), and crude reaction mixture (B) (acrylic esters and dodecanoic esters) using bulk polymerization. | ||
We also performed two experiments for water-dispersive polymerization. Each initial mixture was magnetically stirred at 70 °C for 18 h. The processes were quenched, and the white solids formed were washed until no monomer was observed by GC–FID in the washing solutions.
Table 3 shows the amount of copolymer formed from two starting materials (A1: distilled mixture; B1: crude reaction mixture). The polymerization yield for the two acrylic esters was 70, 2% after 18 h. Both ethyl acrylate and allyl acrylate were incorporated at a high proportion to the polymers formed, although the latter was incorporated in a greater ratio. When the crude reaction mixture was used, ethyl dodecanoate and allyl dodecanoate were almost fully recovered, showing some differences from the equivalent bulk polymerization. These results are in accordance with the elemental analysis of these two copolymers. The C:H ratio for each new polymer did not show any appreciable difference from the respective polymer prepared by bulk polymerization. Again, the FT-IR spectra showed the characteristic IR bands corresponding to the presence of non-conjugated carboxylic esters, alkyl saturated chains, and olefinic bonds. The largest difference between the two polymerization systems was the behaviour of the polymers with respect to toluene. While polymers from bulk polymerization did not swell with toluene, as occurred with all of the solvents tested, the polymers from water-dispersive polymerization did swell.
| Copolymer | Starting material | Initial composition (%) | Monomer conversion (%) | Conversion (%) | Yield (%) |
|---|---|---|---|---|---|
| a Monomer conversion and yield of copolymer based on the acrylate esters. | |||||
| A1 | Ethyl acrylate | 64.3 | 80.7 | 81.4 | 70.2 |
| Allyl acrylate | 35.7 | 82.5 | |||
| B1 | Ethyl acrylate | 28.8 | 36.6 | 52.8a | 50.6a |
| Allyl acrylate | 23.9 | 70.3 | |||
| Ethyl laurate | 22.7 | — | |||
| Allyl laurate | 24.6 | — | |||
The 1H NMR spectra of the copolymers (see ESI†) showed the presence of vinylic protons, although in a lower proportion than expected on the basis of the elemental analysis results. Moreover, the 1H NMR spectra also showed that these vinylic protons corresponded to the allylic residues. These results confirmed the entire polymerization of the acrylic bond and a partial cross-polymerization of the allylic residues.
E-factor
E-factor is one of the metrics proposed for the evaluation of the environmental impact of a given process.57–59Table 4 shows the results obtained for each polymerization process considering that a) the final alkyl ester mixture is recycled to prepare new allyl laurate; b) 80% of the organic solvent used to wash the copolymer is recovered. E-factor ranges from 6.3 to 3.5 showing that bulk polymerizations are preferable in terms of waste production.| Copolymera | A | B | A1 | B1 |
|---|---|---|---|---|
| a Capital letters correspond to the copolymers indicated in Fig. 5 and Table 3. | ||||
| E-factor | 3.8 | 3.5 | 4.1 | 6.3 |
Conclusions
Here, we successfully prepared allyl acrylate by an interesterification reaction using a readily available commercial enzyme in a miniaturized, continuous-flow reactor without solvent. Mixtures of acrylate esters were obtained and polymeric materials were prepared from them using either bulk or water-dispersive polymerization procedures. E-factor ranged from 6.3 to 3.5. The successful synthesis in a miniaturized continuous-flow reactor allows the use of allyl esters obtained from renewable sources as starting materials. The other reagent, ethyl acrylate, can also be obtained from lactic acid. Here, we have demonstrated that miniaturization is convenient for this biocatalytic interesterification because it achieved a similar percentage of ester conversion as a batch reactor. Moreover, the ease in controlling reaction parameters, the minimal use of space needed for the equipment, easy automation, the reproducibility, and the increase in productivity make the flow-reaction system an interesting and advantageous alternative to the batch-reaction system.Acknowledgements
This work was supported in part by a Grant-in-Aid from the Secretaría de Estado de Política Científica y Tecnológica of the Spanish Ministry of Education and Culture (contract grant number: CTQ2009-14699-C02-01). The authors thank the Vicerectorat de Recerca de l'Universitat de Lleida for the grant awarded to Edinson Yara Varón.References
- J. T. Stearn, B. Makower and P. H. Groggins, Ind. Eng. Chem., 1940, 32, 1335–1343 CrossRef CAS.
- Z. Zhang, Y. Qu, S. Wang and J. Wang, Ind. Eng. Chem. Res., 2009, 48, 9083–9089 CrossRef CAS.
- J. M. Lee, D. W. Hwang, Y. K. Hwang, S. B. Halligudi, J. S. Chang and Y. H. Han, Catal. Commun., 2010, 11, 1176–1180 CrossRef CAS.
- J. Zhang, Y. Zhao, M. Pan, X. Feng, W. Ji and C. T. Au, ACS Catal., 2011, 1, 32–41 CrossRef CAS.
- M. H. Jamróz, M. E. Jamroz, J. E. Rode, E. Bednarek and J. C. Dobrowolski, Vib. Spectrosc., 2009, 50, 231–244 CrossRef.
- Y. K. Agarwal, S. D. Kaushik and P. C. Kumar, J. Macromol. Sci., Part A: Pure Appl. Chem., 2007, 44, 877–880 CrossRef CAS.
- F. Sugiyama, K. Satoh and M. Kamigaito, Polym. Prepr. Japan, 2006, 55, 167 Search PubMed.
- Z. M. O. Rzaev, Polym. Int., 2002, 51, 998–1012 CrossRef CAS.
- D. Avci and L. J. Mathias, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 901–907 CrossRef CAS.
- D. Avci and L. J. Mathias, Am. Chem. Soc., Polym. Prep., Div. Polym. Chem., 1998, 39, 484–485 CAS.
- J. Lejnieks, A. Mourran, W. Tillmann, H. Keul and M. Möller, Materials, 2011, 3, 3369–3384 CrossRef.
- L. Liangui, M. Yan, C. Juan, A. Wei and S. Xiuhua, US Pat. CN101993371A, 2011 Search PubMed.
- P. Klan and P. Benovsky, Monatsh. Chem., 1992, 123, 469–471 CrossRef CAS.
- H. Nakagawa, T. Hirabayashi, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2004, 69, 3474–3477 CrossRef CAS.
- M. Nordblad and P. Adlercreutz, Biotechnol. Bioeng., 2008, 99, 1518–1524 CrossRef CAS.
- T. De Diego, A. Manjo, P. Lozano, M. Vaultier and J. L. Iborra, Green Chem., 2011, 13, 444–451 RSC.
- A. B. Hajjar, P. F. Nicks and C. J. Knowles, Biotechnol. Lett., 1990, 12, 825–830 CrossRef CAS.
- S. Warwel, G. Steinke and M. R. Klaas, Biotechnol. Tech., 1996, 10, 283–286 CrossRef CAS.
- M. Nordblad and P. Adlercreutz, J. Biotechnol., 2008, 133, 127–133 CrossRef CAS.
- P. O. Syrén, E. Lindgren, H. W. Hoeffken, C. Branneby, S. Maurer, B. Hauer and K. Hult, J. Mol. Catal. B: Enzym., 2010, 65, 3–10 CrossRef.
- D. Boeckh, B. Hauer and D. Haering, US Pat. 2,006,035,341A1, 2006 Search PubMed.
- W. Paulus, B. Hauer, D. Haring and F. Dietsche, US Pat.2,006,030,013A, 2006 Search PubMed.
- M. Escriba, J. Eras, G. Villorbina, M. Balcells, C. Blanch, N. Barniol and R. Canela, Waste Biomass Valoriz., 2011, 2, 285–290 CrossRef CAS.
- E. Yara-Varón, J. Eras Joli, M. Torres, N. Sala, G. Villorbina, J. J. Méndez and R. Canela-Garayoa, Catal. Today, 2012 DOI:10.1016/j.cattod.2012.02.055.
- A. Hasaninejed, M. R. Kazerooni and A. Zare, Catal. Today, 2012 DOI:10.1016/j.cattod.2012.05.026.
- G. Jas and A. Kirschning, Chem.–Eur. J., 2003, 9, 5708–5723 CrossRef CAS.
- G. Szöllósi, S. Cserényi, K. Balázsik, F. Fülöp and M. Bartók, J. Mol. Catal. A: Chem., 2009, 305, 155–160 CrossRef.
- M. S. Thomsen and B. Nidetzky, Eng. Life Sci., 2008, 8, 40–48 CrossRef CAS.
- P. L. Urban, D. M. Goodall and N. C. Bruce, Biotechnol. Adv., 2006, 24, 42–57 CrossRef CAS.
- L. L. Woodcock, C. Wiles, G. M. Greenway, P. Watts, A. Wells and S. Eyley, Biocatal. Biotransform., 2008, 26, 466–472 CrossRef CAS.
- E. Garcia-Egido, V. Spikmans, S. Y. F. Wong and B. H. Warrington, Lab Chip, 2003, 3, 73–76 RSC.
- M. Escribà, V. Hessel, S. Rothstock, J. Eras, R. Canela and P. Löb, Green Chem., 2011, 13, 1799–1805 RSC.
- J. West, B. Karamata, B. Lillis, J. P. Gleeson, J. Alderman, J. K. Collins, W. Lane, A. Mathewson and H. Berney, Lab Chip, 2002, 2, 224–230 RSC.
- V. Hessel, Chem. Eng. Technol., 2009, 32, 1655–1681 CrossRef CAS.
- F. S. Ekholm, I. M. Mándity, F. Fülöp and R. Leino, Tetrahedron Lett., 2011, 52, 1839–1841 CrossRef CAS.
- S. Ceylan, L. Coutable, J. Wegner and A. Kirschning, Chem.–Eur. J., 2011, 17, 1884–1893 CrossRef CAS.
- I. R. Baxendale, J. Deeley, C.M. Griffiths-Jones, S.V. Ley, S. Saaby and G. K. Tranmer, Chem. Commun., 2006, 24, 2566–2568 RSC.
- A. Mahapatro, B. Kalra, A. Kumar and R. A. Gross, Biomacromolecules, 2003, 4, 544–551 CrossRef CAS.
- M. Escriba, M. Barbut, J. Eras, R. Canela, J. Avilla and M. Balcells, J. Agric. Food Chem., 2009, 57, 4849–4853 CrossRef CAS.
- J. Eras, M. Escriba, G. Villorbina, M. Oromi-Farrus, M. Balcells and R. Canela, Tetrahedron, 2009, 65, 4866–4870 CrossRef CAS.
- O. H. Gonçalves, R. A. F. Machado, P. H. H. de Araújo and J. M. Asua, Polymer, 2009, 50, 375–381 CrossRef.
- B. Yan, C. F. Jewell and S. W. Myers, Tetrahedron, 1998, 54, 11755–11766 CAS.
- G. D. Yadav and K. M. Devi, Biochem. Eng. J., 2002, 10, 93–101 CrossRef CAS.
- E. M. Anderson, K. M. Larsson and O. Kirk, Biocatal. Biotransform., 1997, 16, 181–204 CrossRef.
- E. J. Mammarella and A. C. Rubiolo, Process Biochem., 2009, 44, 183–190 CrossRef CAS.
- S. F. A. Halim, A. H. Kamaruddin and W. J. N. Fernando, Bioresour. Technol., 2009, 100, 710–716 CrossRef CAS.
- E. Séveracd, O. Galy, F. Turond, C. Azzaro Pantele, J. S. Condorete, P. Monsan and A. Marty, Enzyme Microb. Technol., 2011, 48, 61–70 CrossRef.
- X. Xu, S. Balchen, C. E. Høy and J. Adler-Nissen, J. Am. Oil Chem. Soc., 1998, 75, 1573–1579 CrossRef CAS.
- T. H. Rønne, T. Yang, H. Mu, C. Jacobsen and X. Xu, J. Agric. Food Chem., 2005, 53, 5617–5624 CrossRef.
- L. A. Bernardez, L. R. P. de Andrade Lima and P. F. Almeida, BJPG, 2008, 2, 114–121 Search PubMed.
- W. W. Xi and J. H. Xu, Process Biochem., 2005, 40, 2161–2166 CrossRef CAS.
- N. M. Osório, J. H. Gusmão, M. M. da Fonseca and S. Ferreira-Dias, Eur. J. Lipid Sci. Technol., 2005, 107, 455–463 CrossRef.
- N. Arsalani, H. Fattahi and A. A. Entezami, IPJ., 2006, 15, 997–1005 CAS.
- N. Cherifi, A. Issoulie, A. Khoukh, A. Benaboura, M. Save, C. Derailc and L. Billon, Polym. Chem., 2011, 2, 1769–1777 RSC.
- S. Tomoeda, Y. Kitayama, J. Wakamatsu, H. inami, P. B. Zetterlund and M. Okubo, Macromolecules, 2011, 44, 5599–5604 CrossRef CAS.
- M. D. Rikkou and C. S. Patrickios, Prog. Polym. Sci., 2011, 36, 1079–1097 CrossRef CAS.
- K. V. Aken, L. Strekowski and L. Patiny, Beilstein J. Org. Chem., 2006, 2 DOI:10.1186/1860-5397-2-3.
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- M. Sheykhan, Z. R. Ranjbar, A. Morsali and A. Heydari, Green Chem., 2012, 14, 1971–1978 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Figure of flow continuous system, column reactor and 1H NMR spectra for the co-polymers. See DOI: 10.1039/c2ra21503a |
| This journal is © The Royal Society of Chemistry 2012 |