Water-soluble gold nanoparticles stabilized with cationic phosphonium thiolate ligands†
Yon
Ju-Nam
a,
Yu-Su
Chen
b,
Jesus J.
Ojeda
c,
David W.
Allen
b,
Neil A.
Cross
b,
Philip H. E.
Gardiner
b and
Neil
Bricklebank
*b
aMultidisciplinary Nanotechnology Centre, College of Engineering, Swansea University, Singleton Park, Swansea, SA2 8PP, UK
bBiomedical Research Centre, Sheffield Hallam University, City Campus, Sheffield S1 1WB, UK. E-mail: n.bricklebank@shu.ac.uk
cExperimental Techniques Centre, Brunel University, Kingston Lane, Uxbridge, Middlesex, UB8 3PH, UK
First published on 21st September 2012
Abstract
Attachment of cationic groups to the surface of gold nanoparticles (AuNPs) is an attractive proposition for facilitating mitochondria-targeted therapeutics and diagnostics. With this in mind we have prepared and characterised AuNPs functionalised with phosphonium groups derived from either triarylphosphoniopropylthiosulfate zwitterions or ω-thioacetylpropyl(triphenyl)phosphonium salts; organophosphonium cations display remarkable lipophilicity and are readily taken up by cells and are concentrated in the mitochondria. The phosphonium-functionalised AuNPs can be dispersed in water and biological media. Transmission Electron Microscopy reveals the formation of spherical particles with diameters in the range 3–5 nm. The presence of the phosphonioalkylthiolate ligands on the surface of the AuNPs is confirmed by XPS, LDI-TOF-MS, TOF-SIMS and 31P NMR spectroscopy. The phosphonium-AuNPs display excellent stability and preliminary studies indicate that the phosphonioalkylthiolate ligands are slowly oxidised over a period of months to the corresponding phosphonioalkylsulfonate species with a concomitant increase in the particle size, and particle size distribution, of the AuNPs.
1 Introduction
Investigations of gold nanoparticles (AuNPs) with a view to their applications in the fields of biotechnology and biomedical science continue apace.1 This interest stems not least because of their relative ease of preparation, stability, and their apparent non-toxicity. The majority of strategies for the synthesis of AuNPs employ some form of surface coating, usually a ligand, surfactant or polymer, to encapsulate the growing particle, preventing uncontrolled aggregation, providing stability, and imparting functionality. For biological applications the primary requirements are aqueous solubility and compatibility with the immediate biological environment and, for sensor or diagnostic applications, appropriate receptor groups.The most commonly deployed ligands for protecting AuNPs are organic thiolates derived from thiols or disulfides.2 Other common ligands include citrate3 and tertiary phosphines.4 The attachment of ionic ligands to the surface of AuNPs is an attractive proposition for improving the aqueous solubility of the resulting nanoparticles and facilitating cellular uptake5 and biomolecular recognition through non-covalent interactions.6 The most widely studied ligands for the production of anionic-charged AuNPs have been thiolate derivatives of carboxylic acids7 and, for cationic particles, thiolates bearing ammonium groups,8 such as CTAB,9 or other cationic nitrogen systems e.g., ethidium.10 Zwitterionic ligands that have been used to functionalise AuNPs include phosphorylcholine thiolate,11 and a thiolate bearing an ammonium alkyl sulfonate head group.12
Organic phosphonium salts are an important class of lipophilic cations that are readily taken-up by cells and preferentially accumulated in mitochondria.13 This remarkable property has led to the use of phosphonium compounds as anticancer agents,14 intra-cellular transport vectors,15 and as agents for tumour imaging and diagnostics.16 We hypothesised that attachment of phosphonium compounds to the surface of metal nanoparticles would facilitate the absorption of the nanoparticles by cells, making them ideal candidates for mitochondria-targeted pharmaceutical nanotechnology.17 Recent work has shown that incorporation of phosphonium groups into the lipid bilayer of liposomes,18 or onto the surface of dendrimers,19 both of which can act as nanoscale drug delivery systems, facilitates their preferential uptake by mitochondria. These studies demonstrate the potential of phosphonium-functionalised nanocarriers for targeting mitochondria.
With this in mind we have reported the synthesis of a series of phosphonioalkylthiosulfate zwitterions20 and ω-thioacetylalkyl-phosphonium salts21 that behave as masked alkylthiolate ligands, in which the thiolate is ‘protected’ as a thiosulfate or thioacetate group, respectively. Cleavage of the sulfur–sulfur or sulfur–carbon bonds in the thiosulfate and thioacetate species generates thiolate anions in situ leading to water-soluble, cationic, phosphonium-functionalised AuNPs. The mechanisms of the formation of self-assembled monolayers on gold using alkyl thiosulfates (Bunte Salts)22–24 as the precursor ligands has been the subject of recent studies which show that hydrolysis by trace amounts of water facilitates monolayer formation. Similarly, a number of studies have focused on the use of alkyl thioacetates as precursors for the formation of monolayers on gold.25,26
Although there are many studies of the use of organophosphorus ligands, notably phosphines4 and phosphine oxides,27 for passivating the surface of metal nanoparticles, there have been far fewer studies of the use of phosphorus ligands to impart functionality to nanoparticles. The phosphonium ionic liquid trihexyl(tetradecyl)phosphonium bis(2,4,4-trimethylpentyl phosphinate) has been used in the production of luminescent CdSe quantum dots.28 Phosphinophosphonic acids have been used to prepare water soluble Rh and Pt nanoparticles,29 and tris(hydroxymethyl)phosphine (THP)-capped AuNPs have been formed through the reduction of HAuCl4 with tetrakis(hydroxymethyl)phosphonium chloride;30,31 the resulting THP-AuNPs readily form conjugates with DNA.31
In this paper, we report the detailed characterisation of our water-soluble phosphonioalkylthiolate-capped AuNPs in both the solid state and solution using transmission electron microscopy (TEM), mass spectrometry and X-Ray photoelectron spectroscopy (XPS), together with supporting UV-visible and 31P-NMR spectroscopic data. We have also undertaken some preliminary studies into the stability of the compounds with the aim of identifying the fate of the particles over elongated periods of time.
2 Results and discussion
The phosphonium-AuNPs were synthesised from KAuCl4 and triphenylphosphoniopropylthiosulfate (PPTS), tri(p-fluorophenyl)phosphoniopropylthiosulfate (FPPTS) or ω-thioacetylpropyltriphenylphosphonium bromide (PPTA) (Scheme 1) in a two-phase H2O/DCM system employing sodium borohydride as the reducing agent, following the methods described previously.20,21 Samples were purified by extraction with dichloromethane followed by freeze-drying.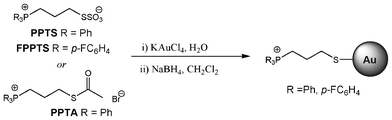 | ||
| Scheme 1 | ||
2.1 TEM studies
The phosphonium-AuNPs were analysed by TEM which showed all the samples to have a spherical shape (Fig. 1). Corresponding statistical analysis of particle size reveals mean diameters of 3.0 ± 1.2 nm (PPTS), 4.9 ± 1.5 nm (PPTA) and 2.7 ± 0.8 (FPPTS). The particles derived from the PPTS and FPPTS zwitterions are similar in size and particle size distribution. However, those prepared with the PPTA salt are larger, and have a broader particle size distribution, than those obtained from the PPTS zwitterion. The differences in size between the AuNPs derived from the PPTS zwitterion and PPTA salt can be explained on the basis of the differing passivation kinetics of the two types of ligand.22–26 The first stage of the synthesis should be adsorption of the ligand on the surface of the growing particles and the thioacetate might be expected to be a more weakly binding ligand than the thiosulfate. Secondly, transformation of the thiosulfate or thioacetate to the thiolate occurs. The mechanism of this step in our system is unclear at present. Recent studies into the formation of monolayers on gold surfaces from alkylthiosulfates show hydrolysis by trace amounts of water present in the solvent to be crucial.23,24 The phosphonium-AuNPs reported here were prepared in a two-phase water/dichloromethane mixture.20,21 However we cannot exclude the possibility that the growing gold nanoparticles and/or the reducing agent are involved in the cleavage of the thiosulfate S–S or thioacetate S–C bonds. It would appear that the cleavage step is slower for the thioacetate than for the thiosulfate and so results in slower monolayer formation.22–26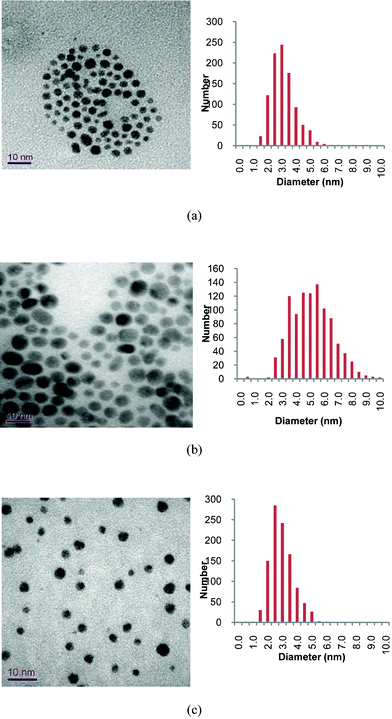 | ||
| Fig. 1 Typical TEM images of phosphonium-AuNPs and associated particle size histograms. Precursor phosphonium ligands are: (a), PPTS; (b) PPTA; (c) FPPTS. | ||
2.2 XPS spectroscopy
In order to determine the elemental composition of the phosphonium-AuNPs, and probe the nature of Au–S bonds, the compounds have been studied by XPS spectroscopy. The data are summarised in Table 1. Wide scan spectra of AuNPs (Fig S1†) were very similar, showing signals due to Au, S, P and C. The XPS spectra, and time-of-flight secondary ion mass spectrometry (TOF-SIMS) (see section 2.3 below) show the presence of chlorine which we postulate is present as chloride that acts as a counter ion to the phosphonium group. In addition, the XPS spectrum of the AuNP capped with the FPPTS compound contained a F(1s) peak, with a binding energy of 687.4 eV, consistent with the presence of fluorine in this sample. The high resolution Au(4f) spectra of all three compounds contain a doublet for Au (4f7/2) and Au (4f5/2) with binding energies of c.a. 84.0 and 87.5 eV, respectively (Fig. 2 and S2†). These values are similar to those reported by Brust (83.8 eV and 87.5 eV),32 and Yee et al. (84.2 eV and 87.85 eV),33 for AuNPs capped with dodecanethiol.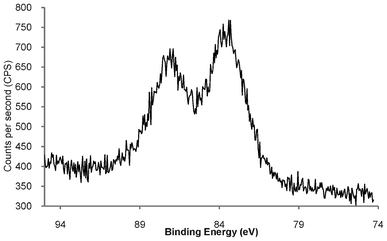 | ||
| Fig. 2 High resolution Au(4f) XPS spectrum of freeze-dried phosphonium-AuNP derived from PPTS zwitterion, showing Au (4f7/2) and Au (4f5/2) doublet with binding energies of 84.0 and 87.5 eV, respectively. | ||
The binding energy of Au 4f7/2 from Au(0) in a metallic gold film is 84 eV, whereas the Au(I) in a gold thiolate has a binding energy of 86 eV and AuNPs that are reported to contain a fraction of their surface atoms in the Au(I) oxidation state show a Au 4f7/2 peak with a binding energy of 84.9 eV.29,34 The Au (4f7/2) observed at 84.0 eV in the phosphonium-AuNPs strongly suggests that the bulk of the gold atoms are in the Au(0) oxidation state.35
Fig. 3 shows the high resolution S(2p) XPS spectrum of the AuNP generated using the PPTS zwitterion, displaying a peak with a binding energy of 162.9 eV, corresponding to S(2p3/2), and a shoulder at ca. 164 eV relating to a S(2p1/2) component.
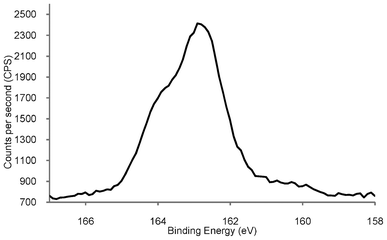 | ||
| Fig. 3 High resolution S(2p) XPS spectrum of phosphonium-AuNPs generated from the PPTS ligand. | ||
The S(2p) XPS spectrum of the phosphonium-AuNPs obtained from the FPPTS ligand contains peaks with very similar binding energies. These values are consistent with a system in which the sulfur species is bound to the surface of the gold as a thiolate, which typically occurs in the range (162.0 to 162.9 eV).36,37 There was no evidence for oxidised sulfur, which shows a peak at ∼167 eV, vide infra. The sulfur XPS spectrum of dodecanethiolate-capped gold nanoparticles is very similar to those reported here, showing an unresolved doublet at 162.9 and 164.2 eV,33 and gold nanoparticles prepared from mercaptoethoxyethanol thiosulfate display S2p binding energies of 162.0 and 164.5 eV.22 The XPS spectrum of the parent PPTS zwitterion (Fig. S3†), displays two S(2p) signals at 161.5 and 166.1 eV, arising from the non-equivalent sulfurs bound to carbon and to oxygen, respectively. This result confirms the cleavage of the sulfur-sulfur bond in the phosphoniopropylthiosulfate zwitterions during the synthesis with the concomitant expulsion of SO32− (or some other sulfur species derived from it), at the ligand/Au interface. The AuNPs produced from the PPTA ligand, display a S(2p3/2) signal at 162.1 eV, which is very similar to that recorded for those derived from the PPTS zwitterion, and identical to the S(2p3/2) peak of AuNPs derived from n-tetradecanethioacetate.26 The spectrum of the precursor PPTA ligand contains a single S(2p3/2) peak with a binding energy of 161.1 eV. This result confirms the cleavage of the thioacetyl moiety from the PPTA ligand during the synthetic process.
2.3 Mass spectrometry analysis
Laser desorption/ionisation time-of-flight mass spectrometry (LDI-TOF-MS) has proved a powerful tool for the analysis of functionalised AuNPs since the surface ligands are efficiently ionised due the ability of the gold particles to absorb at wavelengths commonly used by mass spectrometers.38 We have analysed the phosphonium-AuNPs using LDI-TOF-MS. The spectra of the AuNPs obtained from the PPTS and FPPTS zwitterions (Fig. 4), show fragments corresponding to the phosphoniopropylthiolate ions [Ph3P+(CH2)3S−+H, m/z = 337] and [(p−FC6H4)3P+(CH2)3S−+H, m/z = 391], but do not contain molecular ions for the parent PPTS or FPPTS species. In contrast, the AuNPs obtained from the PPTA salt shows the triphenylphosphoniopropylthiolate fragment [Ph3P+(CH2)3S−+H, m/z = 337] and also a peak corresponding to the parent molecular ion (m/z = 379).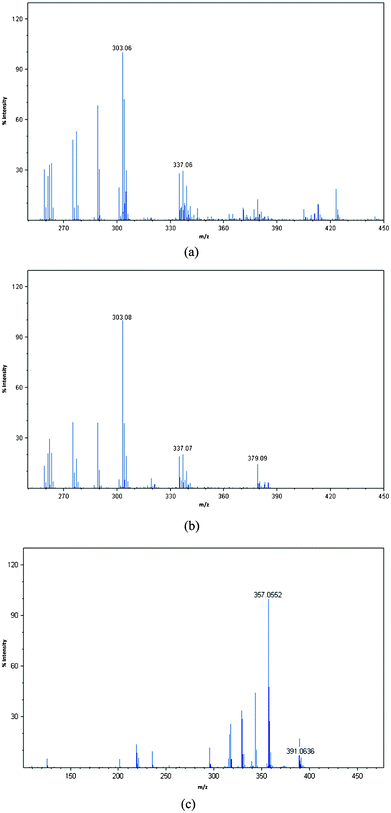 | ||
| Fig. 4 LDI-TOF-MS spectra of phosphonium-AuNPs generated from (a) PPTS, (b) PPTA, (c) FPPTS. | ||
We have also analysed the samples using time-of-flight secondary ion mass spectrometry (TOF-SIMS) which can be used to image the nanoparticle surface.39 During SIMS analysis, a primary ion beam desorbs neutral and ionized molecular fragments from the sample surface. The SIMS spectra, in both positive and negative ion modes (Fig. S4–S6†), showed the presence of C, Cl, S, P, Au and, in the case of the particles prepared using FPPTS, fluorine. The LDI and SIMS mass spectrometry results clearly indicate the presence of the phosphoniopropylthiolate ligands on the surface of the AuNPs. The observation of the molecular ion for the PPTA salt in the LDI-TOF-MS spectrum of the AuNPs prepared from this ligand are in accord with the TEM analysis of this sample which shows particles with a larger diameter than those prepared from the corresponding PPTS zwitterion, consistent with slower, or incomplete, reaction.
2.4 Solution state studies
The freeze-dried particles can be re-suspended in water, methanol, ethanol or DMSO. The UV-visible spectra of the solutions after re-suspension (Fig. S7†) are similar to that of a freshly prepared solution, indicating that the freeze-drying process does not significantly alter the particle size or shape. Furthermore, phosphonium-AuNPs can also be re-dispersed in biological media including phosphate buffered saline (PBS) and Dulbecco's Modified Eagle Medium (DMEM) cell culture medium (Fig. 5). Unlike the nanoparticles re-suspended in deionised water or organic solvents, phosphonium-AuNPs suspended in biological media have a significantly shorter shelf-life and begin to show signs or aggregation after ca. 12 h. This aggregation is presumably promoted by the presence of inorganic ions in the media.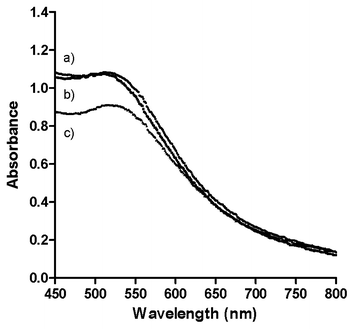 | ||
| Fig. 5 UV-visible spectra of phosphonium-AuNPs prepared using PPTS ligand suspended in biological media. (a) Phosphate Buffered Saline and 10% Foetal Calf Serum; (b) Phenol Red free Dulbecco's Modified Eagle Medium with 10% Foetal Calf Serum; (c) Deionised H2O. | ||
Phosphorus-31 NMR spectra of freeze-dried samples of the phosphonium-AuNPs re-suspended in DMSO show single peaks in the range δ = 23–24 ppm, corresponding to the phosphonium head group of the surface-bound phosphoniopropylthiolate ligand. These values are typical of alkyltriphenylphosphonium salts and indicate that the propylthiolate moiety effectively shields the phosphonium head-group from the electronic effects of the gold core.
2.5 Stability studies
Compared to the total volume of literature on the preparation and characterisation of gold nanoparticles, there are comparatively few investigations into the stability of these species, and most of these discuss the effect of external stimuli, e.g., the effect of light, pH, and the addition of salts.39–45 Yet many of the applications of AuNPs require water-soluble species which are chemically and physically stable for extended periods of time. We have undertaken some preliminary studies into the stability of our phosphonium-AuNPs with the aim of determining the effect of ageing on the particles. Aqueous solutions of phosphonium-AuNPs were stored in the dark under ambient atmosphere and temperature. Samples were analysed by UV-visible spectroscopy at monthly intervals for periods up to 6 months. No change in the position of the surface plasmon band was observed, suggesting that the particle size was the same over this period of time. However, the intensity of the absorption band gradually decreased.We have recorded the XPS spectrum of a freeze-dried sample of the AuNP, prepared using the FPPTS zwitterion, which had been aged for 6 months. The Au(4f) spectrum (Fig. 6a) contains the expected doublet for Au (4f7/2) and Au (4f5/2), with binding energies of 83.8 and 87.0 eV (Table 1), identical to those for a freshly prepared sample of the same AuNP. In contrast, the S(2p3/2) spectrum (Fig. 6b) is markedly different to that of a fresh sample (Fig. 3); the thiolate sulfur signal at 162.9 eV is significantly weaker than for the fresh sample and a second signal with a binding energy of 169.7 eV is observed. It is likely that the latter arises from surface-bound sulfonate species, RSO3−, formed by air oxidation of the thiolate ligands.
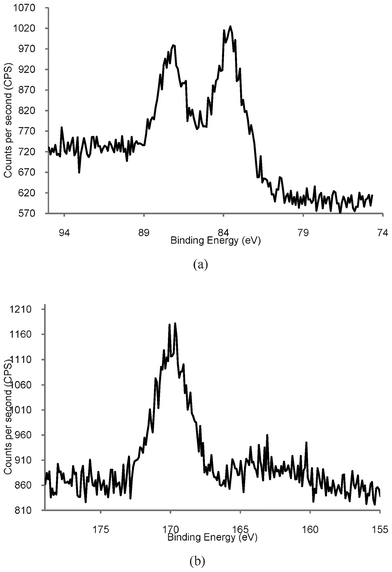 | ||
| Fig. 6 High resolution XPS spectra of Phosphonium-AuNPs prepared using FPPTS after aging for 6 months. (a) Au4f showing expected Au (4f7/2) and Au (4f5/2) doublet. (b) S(2p) showing S(2p3/2) peak at 167 eV. | ||
Although there are very few XPS studies of aged thiolate-capped AuNP samples, there is a much larger body of literature on self-assembled monolayers (SAMs) of organic thiolates on gold surfaces. Organosulfonates display an S(2p) XPS peak with a binding energy of 168 eV,40,41,46–48 which is close to the value reported here. Studies show that air oxidation of thiolate SAMs can occur very rapidly (within hours), is promoted by UV light,40 and that the kinetics of the oxidation are also affected by the nature of the gold surface.47 Rieley et al. proposed a mechanism for the photo-oxidation of thiolate SAMs on Au in which atmospheric O2 penetrates the SAM to reach the Au/thiolate interface and oxidation of the thiolate to sulfonate is mediated by the Au.40 Similarly, Schoenfisch and Pemberton,46 demonstrated that oxidation of alkylthiolate monolayers occurs upon exposure to air, in the absence of light. They concluded that ozone was the likely oxidant and that a number of factors affected the rate of oxidation, including alkyl chain length, monolayer quality, and the nature of the substrate. TEM analysis of our aged sample (Fig. 7) showed an average particle size of 4.5 nm, with some evidence for coalescence with particles up to 10.5 nm in size. These observations are supported by data recorded for phosphonium-AuNPs obtained from the ω-thioacetylhexyl(triphenyl)-phosphonium bromide ligand,21 which had been aged for 6 months. The sulfur XPS spectrum of this sample displayed a S(2p3/2) peak with a binding energy of 167 eV and scanning-tunnelling electron microscopy showed a mixture of different sized particles.49
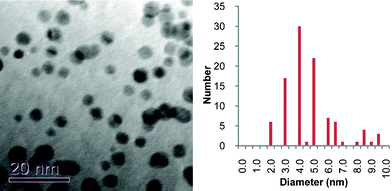 | ||
| Fig. 7 TEM micrograph and particle size histogram of freeze-dried gold nanoparticles prepared from FPPTS zwitterion that had been aged for 6 months. | ||
Organic sulfonate species have a lower affinity for gold surfaces than the corresponding un-oxidised thiolate, and are desorbed from the surface of the gold substrate. We speculate that the phosphonioalkylthiolate ligands undergo a similar oxidation process to the alkylthiolate SAMs.40,46 However, whereas oxidation of thiolate-coated SAMs can occur very quickly, the oxidation of triarylphosphonioalkylthiolate appears to occur much more slowly. A possible explanation is that the bulky triarylphosphonium head groups may shield the Au/thiolate interface from O2 and so oxidation to a phosphonioalkylsulfonate occurs over a longer timescale. The phosphonioalkylsulfonates may then desorb from the surface of the AuNP and once a significant number of ligands have oxidised, the AuNPs begin to coalesce and aggregate into larger clusters.
3 Conclusion
In this paper we have presented data which show that triarylphosphoniopropylthiosulfate zwitterions and ω-thioacetylpropyl (triphenyl)phosphonium salts can be used to prepare cationic, water-soluble AuNPs with mean core sizes in the range 3–5 nm. Comparison of phosphonium-AuNPs produced from the PPTS zwitterions with those derived from the corresponding PPTA salt shows that, although the final particles have the same Au–S ligation, the PPTA salt produce slightly larger AuNPs than the PPTS zwitterion. Initial studies indicate that phosphonium-AuNPs are stable for periods of several months before significant degradation begins to take place. XPS and TEM analysis of AuNPs derived from the FPPTS zwitterion after 6 months revealed the presence of oxidised tris(p-fluorophenyl)phosphoniopropylthiosulfonate species. We propose that the oxidation of the phosphonioalkylthiolate ligands leads to the gradual coalescence and aggregation of the AuNPs.The presence of the phosphonium ligands on the surface of the AuNPs is potentially of great interest for biomedical applications as a result of the high affinity of the triphenylphosphonium moiety for biological systems, particularly mitochondria. Biological applications of AuNPs require materials that can be dispersed in aqueous solution or biological media and we have demonstrated this behaviour for our compounds. We are currently undertaking detailed studies of the cellular biology of the phosphonioalkylthiolate-capped AuNPs.
4 Materials and methods
4.1 Reagents
Chemicals and solvents were purchased from Sigma-Aldrich or Fisher Scientific Ltd and used as received. Samples of triphenylphosphoniopropylthiosulfate (PPTS), tri(p-fluorophenyl)phosphoniopropylthiosulfate (FPPTS) and ω-thioacetylpropyltriphenylphosphonium bromide (PPTA), and the corresponding gold nanoparticles were prepared following the protocols reported previously.20,214.2 Characterisation of nanoparticles
XPS measurements were made on a VG Escalab 210 Photoelectron Spectrometer. The X-ray source was a non-monochromated Al Kα source (1486.6 eV), operated with an X-ray emission current of 20 mA and an anode high tension (acceleration voltage) of 12 kV. The freeze-dried sample was placed on a standard sample stud employing double sided adhesive tape and the take-off angle was fixed at 90° relative to the sample plane. The area corresponding to each acquisition was of 0.79 mm2. Each analysis consisted of a wide survey scan (pass energy 50 eV, 1.0 eV step size) and high-resolution scans (pass energy 50 eV, 0.05 eV step size) for component speciation. The binding energy scale of the instrument was calibrated using the Au 4f5/2 (83.9 eV), Cu 2p3/2 (932.7 eV) and Ag 3d5/2 (368.27 eV) lines of cleaned gold, copper and silver standards from the National Physical Laboratory (NPL), UK. The software CasaXPS 2.3.15 was used to fit the XPS spectra peaks, and the binding energies obtained in the XPS spectra were corrected for specimen charging by referencing the C 1s to 284.6 eV.TEM micrographs: Air-drying of a suspension droplet was used as the TEM sample preparation method. A Pasteur pipette was used to place a drop of AuNPs, suspended in ethanol, on a Holey carbon coated copper grid. The grids were left to dry in air at room temperature. A Jeol 2000FX transmission electron microscope set at 100 KV was used for the evaluation. Size distribution was obtained over 1000 AuNPs using Abel imaging software.
LDI-TOF-MS experiments were performed using an Applied Biosystems/MDS Sciex hybrid quadrapole time-of-flight mass spectrometer (Q-Star Pulsar-i) with an orthogonal MALDI ion source (Applied Biosystems, Foster City, California, USA) and a high repetition Neodymium-doped yttrium vandate (Nd: YVO4 laser (5 KHz) (Elforlight Ltd, Daventry, Northamptonshire, UK).
Secondary ion mapping studies were performed with a Kore Technology Ltd. time-of-flight secondary ion mass spectrometry (TOF-SIMS) instrument, using a 25 keV Indium primary ion source (FEI Liquid Metal Ion Gun) operating at 1 μA current. Secondary ions were analysed in a reflectron mass spectrometer and detected with a dual microchannel plate assembly. Flight times were recorded with a 0.5 ns time-to-digital converter. Spectra were taken from an area of approximately 250 micrometres square. The freeze-dried sample was placed on a standard sample stud employing double-sided adhesive tape sample. Calibration of the mass spectra was established by defining a common series of CxHy peaks with a known mass using the mass calibration function in the instrument's software.
UV-Vis spectra were recorded on a Jenway 6715 UV/Vis spectrophotometer (Bibby Scientific Limited, Staffordshire, UK) in the wavelength range from 450–800 nm in a UV-quartz cuvette (10 mm optical path).
NMR Spectra were recorded using a Brucker AVANCE III (400 MHz) spectrometer.
Acknowledgements
We are grateful to Sheffield Hallam University for financial support and the provision of bursaries to YSC and YJN. Ms. Nita Verma (Experimental Techniques Centre, Brunel University) is thanked for the provision of the TEM micrographs and particle size distribution.References
- (a) For recent reviews see: Y.-C. Yeh, B. Creran and V. M. Rotello, Nanoscale, 2012, 4, 1871–1880 RSC; (b) E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC; (c) H. Jans and Q. Huo, Chem. Soc. Rev., 2012, 41, 2849–2866 RSC; (d) T. L. Doane and C. Burda, Chem. Soc. Rev., 2012, 41, 2885–2911 RSC; (e) R. R. Arvizo, S. Bhattacharyya, R. A. Kudgus, K. Giri, R. Bhattacharya and P. Mukherjee, Chem. Soc. Rev., 2012, 41, 2943–2970 RSC; (f) D. F. Moyano and V. M. Rotello, Langmuir, 2011, 27, 10376–10385 CrossRef CAS.
- D. Witt, R. Klajn, P. Barski and B. A. Grzybowski, Current Organic Synthesis, 2004, 8, 1763–1797 CAS.
- (a) J. Turkevitch, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC; (b) G. Frens, Nature: Physical Science, 1973, 241, 20–22 CAS.
- P. M. Shem, R. Sardar and J. S. Shumaker-Parry, Langmuir, 2009, 25, 13279–13283 CrossRef CAS.
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS.
- B. A. Armitage, in DNA Binders and Related Subjects, Springer, 2005, 253, 55 Search PubMed.
- (a) Y. Song, T. Huang and R. W. Murray, J. Am. Chem. Soc., 2003, 125, 11694–11701 CrossRef CAS; (b) R. S. Ingram, M. J. Hostetler and R. W. Murray, J. Am. Chem. Soc., 1997, 119, 9175–9178 CrossRef CAS.
- (a) C. A. Fields-Zinna, R. Sardar, C. A. Beasley and R. W. Murray, J. Am. Chem. Soc., 2009, 131, 16266–16271 CrossRef CAS; (b) S. S. Agasti, C.-C. You, P. Arumugam and V. M. Rotello, J. Mater. Chem., 2008, 18, 70–73 RSC; (c) C. M. McIntosh, E. A. Esposito, A. K. Boal, J. M. Simard, C. T. Martin and V. M. Rotello, J. Am. Chem. Soc., 2001, 123, 7626–7629 CrossRef CAS.
- L. Vigderman, P. Manna and E. R. Zubarev, Angew. Chem., Int. Ed., 2012, 51, 636–641 CrossRef CAS.
- G. Wang, J. Zhang and R. W. Murray, Anal. Chem., 2002, 74, 4320–4327 CrossRef CAS.
- Q. Jin, J.-P. Xu, J. Ji and J.-C. Shen, Chem. Commun., 2008, 3058–3060 RSC.
- L. L. Rouhana, J. A. Jaber and J. B. Schlenoff, Langmuir, 2007, 23, 12799–12801 CrossRef CAS.
- (a) M. F. Ross, G. F. Kelso, F. H. Blaikie, A. M. James, H. M. Cocheme, A. Filipovska, T. Da Ros, T. R. Hurd, R. A. J. Smith and M.P. Murphy, Biochemistry (Moscow), 2005, 70, 222–230 CrossRef CAS; (b) R. A. J. Smith, C. M. Porteous, A. M. Gane and M. P. Murphy, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5407–5412 CrossRef CAS.
- (a) M. Millard, D. Pathania, Y. Shabaik, L. Taheri, J. Deng and N. Neamati, PLoS One, 2010, 5, e13131 Search PubMed; (b) K. L. Bergeron, E. L. Murphy, O. Majofodun, L. D. Munoz, C. C. Williams Jr. and K. H. Almeida, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2009, 673, 141–148 CrossRef CAS; (c) D. C. Rideout, T. Calogeropoulou, J. S. Jaworski, R. Dagnino Jr. and M. R. McCarthy, Anti-Cancer Drug Design, 1989, 4, 265–280 CAS.
- (a) A. T. Hoye, J. E. Davoren, P. Wipf, M. P. Fink and V. E Kagan, Acc. Chem. Res., 2008, 41, 87–97 CrossRef CAS; (b) M. P. Murphy and R. J. A. Smith, Annu. Rev. Pharmacol., 2007, 47, 629–656 CrossRef CAS; (c) M. F. Ross, T. Da Ros, F. H. Blaikie, T. A. Prime, C. M. Porteous, I. I. Severina, V. P. Skulachev, H. G. Kjaergaard, R. A. J. Smith and M. P. Murphy, Biochem. J., 2006, 400, 199–208 CrossRef CAS.
- (a) Y. Zhou and S. Liu, Bioconjugate Chem., 2011, 22, 1459–1472 CrossRef CAS; (b) J. A. Ioppolo, J. K. Clegg and L. M. Rendina, Dalton Trans., 2007, 1982–1985 RSC; (c) Y.-S. Kim, C.-T. Yang, J. Wang, L. Wang, Z.-B. Li, X. Chen and S. Liu, J. Med. Chem., 2008, 51, 2971–2984 CrossRef CAS; (d) C.-T. Yang, Y. Li and S. Liu, Inorg. Chem., 2007, 46, 8988–8997 CrossRef CAS; (e) I. Madar, H. Ravert, B. Nelkin, M. Abro, M. Pomper, R. Dannals and J. J. Frost, Eur. J. Nucl. Med. Mol. Imaging, 2007, 34, 2057–2065 CrossRef CAS.
- (a) see for example, Organelle-Specific Pharmaceutical Nanotechnology, ed. V. Weissig and G. G. M. D'Souza, John Wiley, New York, 2010 Search PubMed; (b) V. Weissig, Pharm. Res., 2011, 28, 2657–2668 CrossRef CAS.
- (a) S. V. Boddapati, G. G. M. D'Souza, S. Erdogan, V. P. Torchilin and V. Weissig, Nano Lett., 2008, 8, 2559–2563 CrossRef CAS; (b) S. Biswas, N. S. Dodwadkar, P. P. Deshpande and V. P. Torchilin, J. Controlled Release, 2012, 159, 393–402 CrossRef CAS; (c) S. V. Boddapati, P. Tongcharoensirikul, R. N. Hanson, G. G. M. D'Souza, V. P. Torchilin and V. Weissig, J. Liposome Research, 2005, 15, 49–58 CAS.
- S. Biswas, N. S. Dodwadkar, A. Piroyan and V. P. Torchilin, Biomaterials, 2012, 33, 4773–4782 CrossRef CAS.
- Y. Ju-Nam, N. Bricklebank, D. W. Allen, P. H. E. Gardiner, M. E. Light and M. B. Hursthouse, Org. Biomol. Chem., 2006, 4, 4345–4351 CAS.
- Y. Ju-Nam, D. W. Allen, P. H. E. Gardiner and N. Bricklebank, J. Organomet. Chem., 2008, 693, 3504–3508 CrossRef CAS.
- S. E. Lohse, J. A. Dahl and J. E. Hutchinson, Langmuir, 2010, 26, 7504–7511 CrossRef CAS.
- R. J. Fealy, S. R. Ackerman and G. S. Ferguson, Langmuir, 2011, 27, 5371–5376 CrossRef CAS.
- R. G. Pillai and M. S. Freund, Langmuir, 2011, 27, 9028–9033 CrossRef CAS.
- A. Singh, D. H. Dahanayaka, A. Biswas, L. A. Bumm and R. A. Haltermann, Langmuir, 2010, 26, 13221–13226 CrossRef CAS.
- S. Zhang, G. Leem and T. R. Lee, Langmuir, 2009, 25, 13855–13860 CrossRef CAS.
- (a) W. H. Binder, R. Sachsenhofer, C. J. Straif and R. Zirbs, J. Mater. Chem., 2007, 17, 2125–2132 RSC; (b) M. Green and P. O'Brien, Chem. Commun., 2000, 183–184 RSC.
- M. Green, P. Rahman and D. Smyth-Boyle, Chem. Commun., 2007, 574–576 RSC.
- (a) M. Richter, A. Karschin, B. Spingler, P. C. Kunz, W. Meyer-Zaika and W. Kläui, Dalton Trans., 2012, 41, 3407–3413 RSC; (b) J. Glöckler, S. Klützke, W. Meyer-Zaika, A. Reller, F. J. García-García, H.-H. Strehblow, P. Keller, E. Rentschler and W. Kläui, Angew. Chem., Int. Ed., 2007, 46, 1164–1167 CrossRef.
- L. Shang, R. M. Dörlich, S. Brandholt, R. Schneider, V. Trouillet, M. Bruns, D. Gerthsen and G. U. Nienhaus, Nanoscale, 2011, 3, 2009–2014 RSC.
- (a) D. G. Duff, A. Baiker and P. P. Edwards, Langmuir, 1993, 9, 2301–2309 CrossRef CAS; (b) W. Vogel, D. G. Duff and A. Baiker, Langmuir, 1995, 11, 401–404 CrossRef CAS; (c) O. Harnack, W. E. Ford, A. Yasuda and J. M. Wessels, Nano Lett., 2002, 2, 919–923 CrossRef CAS.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- C. K. Yee, A. Ulman, J. D. Ruiz, A. Parikh, H. White and M. Rafailovich, Langmuir, 2003, 19, 9450–9458 CrossRef CAS.
- A. McNeillie, D. H. Brown, W. E. Smith, M. Gibson and L. Watson, J. Chem. Soc., Dalton Trans., 1980, 767–769 RSC.
- M. J. Hostetler, J. E. Wingate, C. J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J.Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17–30 CrossRef CAS.
- M.-C. Bourg, A. Badia and R. B. Lennox, J. Phys. Chem. B, 2000, 104, 6562–6567 CrossRef CAS.
- C. D. Bain, H. A. Biebuyck and G. M. Whitesides, Langmuir, 1989, 5, 723–727 CrossRef CAS.
- (a) Z.-J. Zhu, P. S. Ghosh, O. R. Miranda, R. W. Vachet and V. M. Rotello, J. Am. Chem. Soc., 2008, 130, 14139–14143 CrossRef CAS; (b) B. Yan, Z.-J. Zhu, O. R. Miranda, A. Chompoosor, V.M. Rotello and R.W. Vachet, Anal. Bioanal. Chem., 2010, 396, 1025–1035 CrossRef CAS.
- D. R. Baer, D. J. Gaspar, P. Nachimuthu, S. D. Techane and D. G. Castner, Anal. Bioanal. Chem., 2010, 396, 983–1002 CrossRef CAS.
- H. Rieley, G. K. Kendall, F. W. Zemicael, T. L. Smith and S. Yang, Langmuir, 1998, 14, 5147–5153 CrossRef CAS.
- G. Mani, D. M. Johnson, D. Marton, V. L. Dougherty, M. D. Feldman, D. Patel, A. A. Ayon and C. M. Agrawal, Langmuir, 2008, 24, 6774–6784 CrossRef CAS.
- N. Satoh, H. Hasegawa, K. Tsujii and K. Kimura, J. Phys. Chem., 1994, 98, 2143–2147 CrossRef CAS.
- I. Ojeda-Jiménez and V. Puntes, J. Am. Chem. Soc., 2009, 131, 13320–13327 CrossRef.
- D. I. Gittins and F. Caruso, Angew. Chem., Int. Ed., 2001, 40, 3001–3004 CrossRef CAS.
- Y.-S. Shon, S. Chuc and P. Voundi, Colloids Surf., A, 2009, 352, 12–17 CrossRef CAS.
- M. H Schoenfisch and J. E. Pemberton, J. Am. Chem. Soc., 1998, 120, 4502–4513 CrossRef.
- M.-T. Lee, C.-C. Hsueh, M. S. Freund and G. S. Ferguson, Langmuir, 1998, 14, 6419–6423 CrossRef CAS.
- N. T. Flynn, T. N. T. Tran, M. J. Cima and R. Langer, Langmuir, 2003, 19, 10909–10915 CrossRef CAS.
- Y. Ju Nam, PhD Thesis, Sheffield Hallam University, 2007 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: supplementary figures. See DOI: 10.1039/c2ra21421k |
| This journal is © The Royal Society of Chemistry 2012 |