Emerging π-stacked dynamic nanostructured library†
Dnyaneshwar B.
Rasale
,
Indrajit
Maity
and
Apurba K.
Das
*
Department of Chemistry, Indian Institute of Technology Indore, IET-DAVV Campus Khandwa Road, Indore 452017, India. E-mail: apurba.das@iiti.ac.in; Fax: + 91 731 2366382; Tel: +91-731-2438-738
First published on 29th August 2012
Abstract
We describe a general strategy to control the state of molecular self-assembly under thermodynamic control in which gelation facilitates formation of a predominating π-stacked nanostructured product.
Self-assembly1 plays a vital role in constructing complex functional materials for applications in biosensing,2 3D matrices for cell culture,3 drug delivery4 and wound healing5 treatment. Various small molecules, including amino acids,6 carbohydrates7 and antibiotics,8 have been used in the development of supramolecular hydrogels. Peptide self-assembly leading to supramolecular hydrogels has been reported in response to external stimuli including pH, ionic strength, temperature, light and enzyme-catalyzed reactions.9 Enzyme catalyzed peptide hydrogels have shown promise in biomedical applications.10 It has been reported that enzymatic conversion promotes the formation of more ordered nanostructures in supramolecular hydrogels.11 Xu and co-workers reported that intracellular enzymatic formation of nanofibers results in hydrogelation.12
Dynamic covalent chemistry exploits the reversibility of chemical reactions for the generation of library molecules under thermodynamic control. In recent years, dynamic combinatorial libraries (DCLs)13 are envisaged to build precise molecular architectures. Sanders et al. reported one of the most successful hydrazone exchange reactions for the generation of a dynamic combinatorial library.14 In addition to host–guest relationships, covalent and non-covalent interactions alter the distribution of library members. Lehn and co-worker developed constitutional dynamic libraries based on supramolecular and reversible connections which involve self-organization and component selection to generate dynamic hydrogels.15 Ulijn et al. have shown the construction of a dynamic library of building blocks of self-assembled hydrogelators.16 Few principles need to be designed for the successful generation of dynamic libraries where self-assembly facilitates towards the most preferred component. Enzyme catalyzed peptide hydrolysis reactions are close to equilibrium in aqueous medium and are thermodynamically favoured.17 Our objective is to exploit reversible breaking/making of peptide bonds and non-covalent interactions that lead to the formation of a single predominating product among the library members. The self-assembly of molecules is based on the formation of hydrogen bonding as well as π–π stacking interactions of highly conjugated aromatic moieties.18 Here, we report the generation of a dynamic library of small peptides with an N-terminal aromatic protecting group i.e. naphthalene-2-methoxycarbonyl (Nmoc) that self-assemble to form self-supporting hydrogels and ultimately lead to a nanostructured predominating product via hydrogen bonding and π-stacking interactions (Fig. 1).
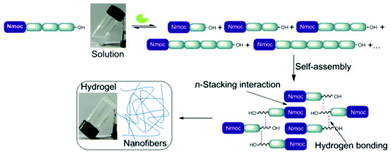 | ||
| Fig. 1 Schematic representation showing enzymatic hydrolysis/reverse hydrolysis, self-assembly of a single predominating product and formation of a self-supporting hydrogel. | ||
DCLs are established when library components continuously exchange themselves via reversible reactions under thermodynamic control. A viscous solution of Nmoc-VVV 1 was observed in a pH dependent manner at pH 6.5–7 at a concentration of 0.9 wt% but it could not form gel rather than a viscous solution. Non-specific endoprotease enzyme thermolysin was added to the solution of Nmoc-VVV and was left at room temperature for about 3 h, resulting in the formation of a self-supporting rigid hydrogel. The hydrogel remained stable for two months. Thermolysin was chosen as the endoprotease to bring about peptide hydrolysis and reverse hydrolysis (Scheme 1) of peptide 1 leading to the formation of a predominating product Nmoc-V at pH 7 driven by π-stacking interactions of aromatic Nmoc groups. The HPLC results indicated that 87% of 1 was converted to Nmoc-V after 48 h (Fig. 2a) and that self-assembly of this newly generated species leads to a self-supporting supramolecular hydrogel. Based on the above DCL concept, the synthesized compound Nmoc-FFF 2 was also tested for hydrogelation. Nmoc-FFF 2 was dispersed and solubilised in water at a concentration of 1.2 wt% by the dropwise addition of 0.5 M NaOH, then gradually decreased to pH 7 by the slow addition of 0.1 M HCl. This system turned into a self-supporting hydrogel. Compound 1 itself self-assembles in water and forms a hydrogel upon treatment with enzyme and generates a library of components with the single predominating product Nmoc-V. However, compound 2 showed exactly reverse behaviour to 1 on treatment with thermolysin. It forms a library of four components with an uneven distribution and turns to liquid under similar conditions (Fig. S12, ESI†). All the newly synthesized products were characterized by reverse phase HPLC and ESI-MS (Fig. S14–S16, ESI†). The hydrogel 1 starts breaking slowly when the pH is lowered below 5. It is entirely precipitated out at pH 4. At higher pH (>8) the gel also starts breaking slowly and at pH 11 the gel to sol transition occurs completely.
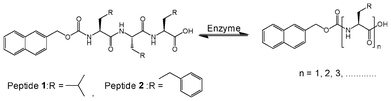 | ||
| Scheme 1 Reversible amide bond synthesis/hydrolysis reaction in which peptide derivatives are formed from Nmoc-tripeptides 1 and 2. | ||
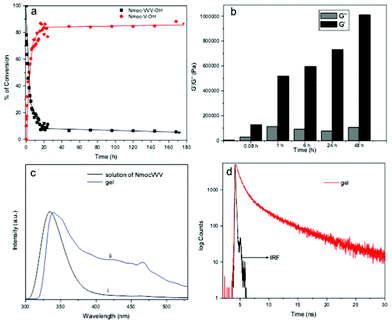 | ||
| Fig. 2 (a) Percentage of Nmoc-V conversion as a function of time followed by HPLC. (b) Comparison between the storage modulus (G′) and loss modulus (G′′) at a particular point of angular frequency (10.7 s−1) with the course of reaction time at a constant strain of 0.1%. (c) Normalized fluorescence spectra taken during the self-assembly process: (i) solution of Nmoc-VVV prior to enzyme addition and (ii) gel of Nmoc-V after enzyme addition (λex = 265 nm). (d) Emission decay curves of a gel Nmoc-VVV/Nmoc-V system monitored at 463 nm after a day (IRF: instrument response function). | ||
Nmoc-amino acids (valine, V; phenylalanine, F) with a fourfold excess of nucleophiles (VV, FF dipeptides) were solubilised in water and thermolysin was added to the reaction mixture at pH 8.16 Newly generated product was not obtained from the Nmoc-V/VV system. 72% Nmoc-FFF (Fig. S16, ESI†) was formed from the Nmoc-F/FF system at 24 h and it turned into a hydrogel within 1 h of enzyme addition. Four fold excess of nucleophile is required for reverse hydrolysis reactions under similar conditions to peptide hydrolysis reactions. Nmoc-V is the predominating product obtained from parent Nmoc-VVV using enzyme catalyzed peptide hydrolysis reaction. These results clearly demonstrate that enzyme-catalyzed self-assembly (hydrogelation) facilitates the formation of the most stable predominating component.
The viscoelastic properties of the hydrogel Nmoc-VVV/Nmoc-V system were characterized using oscillatory rheology. A rigid strong hydrogel has a storage modulus (G′) value that exceeds the loss modulus (G′′) value by an order of magnitude.19 Dynamic frequency sweep testing was carried out to study the hydrogel formation on different time scales (Fig. S18, S19, S20 ESI†). G′ overlaps with G′′ (Fig. 2b) suggesting that the viscous solution of Nmoc-VVV is not fairly strong prior to enzyme addition. Thermolysin was added to the solution of 0.9 wt% of peptide Nmoc-VVV at pH 7 for rheological studies. However G′ and G′′ increased with time, the value of G′ dominating over G′′ about 5 min after addition of thermolysin (Fig. 2b). As shown in Fig. 2b, G′ was much higher than G′′ after about 1 h of reaction. After about 2 days of enzyme reaction the G′ value for the hydrogel is almost 20 times that of G′′, indicating the extensive 3D network in the hydrogel.
Fluorescence emission spectra were recorded to gain more insight into the molecular arrangement of the gelator molecules in the gel phase. As peptide 1 formed a gel only upon enzyme addition, the emission peak originating from the naphthalene double ring centered at 334 nm (in solution) was red shifted to 340 nm (gel phase) (Fig. 2c). The hydrogel formed by self-assembly of a predominating library member Nmoc-V derived from peptide Nmoc-VVV which forms efficient π–π stacking interactions in the gel phase medium. An emission peak centered at 463 nm suggests excimer formation at a higher order aggregate state (Fig. S3†). A time resolved fluorescence study was acquired to investigate the higher order aggregated states of the fluorophore groups (Nmoc) of the Nmoc-V hydrogel. Time resolved fluorometry allows very keen discrimination between fluorophore species in different environments by contributing individual emissions at the same wavelength. The fluorescence decay time of the Nmoc-V hydrogel was measured at an excitation wavelength of 376 nm and the emission was monitored at 463 nm. Tri-exponential decay was fitted to measure the lifetime of Nmoc-V hydrogel at 1 day after enzyme addition (Fig. 2d). The average lifetime was 0.7755 ns of Nmoc-VVV prior to enzyme addition. The average lifetime of 1.108 ns of the Nmoc-V hydrogel was observed a day after enzyme addition at room temperature (Tables S1 and S2†). This value, showing the fluorescence lifetime of the naphthalene groups of hydrogel samples with time scale (Table S3†), indicates a more dense aggregated nanofibrous network in the hydrogel.20
Field-emission scanning electron microscopy (SEM) was used to reveal the self-assembled nanostructures of the hydrogel in a time dependent manner that relate to the self-assembly kinetics of the system. As shown in Fig. 3, the hydrogelator Nmoc-V self-assembled into nanofibers that cross-linked to form a fibrous network. The image (Fig. 3a) shows the dense mass of Nmoc-VVV prior to enzyme addition when the conversion of gelator was zero. However, there was an extreme change in morphology of the hydrogelator Nmoc-V after enzyme addition. Fig. 3b shows a highly dense mass of gelator because of the inefficient self-assembly of the hydrogelator Nmoc-V when the conversion of Nmoc-V was only 10% up to 5 min after enzyme addition. As mentioned earlier, library member Nmoc-V forms a hydrogel but not its parent molecule Nmoc-VVV. The removal of two hydrophobic units from peptide Nmoc-VVV in response to enzyme hydrolysis forms a delicate balance of molecular interaction in the self-assembled nanofiber networks, which results in viscous sol–gel transition. The SEM image in Fig. 3c reveals short entangled nanofibers with an average width of 30 nm at 1 h after enzyme addition. Fig. 3d and 3e show a dense, aggregated nanofibrous network which appears quite random and exhibits an ordered microstructure. However, the sample (Fig. 3f) with one month long enzyme reaction interestingly exhibits quite elongated and thick nanofibers. HPLC analysis showed that the conversion of Nmoc-V after 6 h and 24 h was 67% and 76% respectively. These network structures of nanofibers21 of a preferred library member are responsible for stable hydrogel formation.
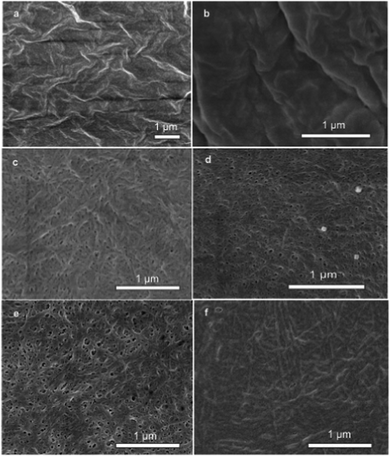 | ||
| Fig. 3 FE-SEM images of the Nmoc-VVV/Nmoc-V system. SEM images of (a) Nmoc-VVV before addition of enzyme and after enzyme addition at (b) 5 min, (c) 1 h, (d) 6 h, (e) 1 day and (f) 1 month. (b) to (f) show the change of nanostructural morphology with subsequent formation of Nmoc-V leading to formation of a self-supporting hydrogel. | ||
In summary, we have demonstrated a new kind of dynamic supramolecular hydrogel by introducing an N-terminus protecting aromatic moiety i.e. the naphthalene-2-methoxycarbonyl (Nmoc) group. Enzyme catalyzed hydrogelation leads to the formation of a dynamic library. The dynamic peptide library generates a very simple nanostructured predominating product that self-assembles through π–π stacking interactions. A real-time morphological change was also described in this communication, which will be a powerful methodology for making biomaterials.
Experimental section
Preparation of hydrogel
Nmoc-VVV (20 mg, 20 mmol L−1) was dispersed in 2 mL water. The pH of the peptide–water mixture was first increased to pH 10 by addition of 0.5 M NaOH, thereby solubilizing the peptide, and then gradually taken back to pH 6.5–7 by slow addition of 0.1 M HCl. Nmoc-VVV could not form a gel. 1 mg (∼40 U mg−1) thermolysin was added to the reaction vial. It turned to strong a gel within 1 h of reaction time and was stable for two months. Nmoc-FFF solutions were formed in a similar manner.20 mmol Nmoc-amino acids (valine, V; phenylalanine, F) with a fourfold excess of nucleophile (VV, FF dipeptides) were solubilised in 2 mL water by the dropwise addition of 0.5 M NaOH and the pH was adjusted to 8 by addition of 0.1 M HCl. Thermolysin (1 mg) was added to the reaction mixture and product formation was detected using HPLC.
HPLC analysis
A Dionex HPLC-Ultimate 3000 (reverse phase high performance liquid chromatography) pump was used to analyze library components of Nmoc-VVV/Nmoc-V and Nmoc-FFF/Nmoc-F systems. The sample preparation involved mixing 100 μL of gel with acetonitrile–water (900 μL, 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture) containing 0.1% trifluoroacetic acid. The samples were then filtered through a 0.45 μm syringe filter (Whatman, 150 units, 13 mm diameter, 2.7 mm pore size) prior to injection. A 20 μL of sample was injected into a Dionex Acclaim ® 120 C 18 column of 250 mm length with an internal diameter of 4.6 mm and 5 μm fused silica particles at a flow rate of 1 mL min−1 (linear gradient of 40% v/v acetonitrile in water for 4 min, gradually rising to 100% v/v acetonitrile in water at 35 min). This concentration was kept constant until 40 min when the gradient was decreased to 40% (v/v) acetonitrile in water at 42 min. The library components of Nmoc-VVV/Nmoc-V and Nmoc-FFF/Nmoc-F systems were identified by using an Ultimate 3000 RS variable wavelength detector at 280 nm. The peak retention times and peak areas were compared with known standards.
:50 mixture) containing 0.1% trifluoroacetic acid. The samples were then filtered through a 0.45 μm syringe filter (Whatman, 150 units, 13 mm diameter, 2.7 mm pore size) prior to injection. A 20 μL of sample was injected into a Dionex Acclaim ® 120 C 18 column of 250 mm length with an internal diameter of 4.6 mm and 5 μm fused silica particles at a flow rate of 1 mL min−1 (linear gradient of 40% v/v acetonitrile in water for 4 min, gradually rising to 100% v/v acetonitrile in water at 35 min). This concentration was kept constant until 40 min when the gradient was decreased to 40% (v/v) acetonitrile in water at 42 min. The library components of Nmoc-VVV/Nmoc-V and Nmoc-FFF/Nmoc-F systems were identified by using an Ultimate 3000 RS variable wavelength detector at 280 nm. The peak retention times and peak areas were compared with known standards.
Characterization
The morphologies of the peptide nanofibers were characterized using a field-gun scanning electron microscope (Jeol JSM-7600F). 2 mL samples were prepared in a 1 cm2 quartz cuvette and emission spectra were measured on FluoroMax-4P fluorimeter from Horiba (Model: FM-100) and the excitation wavelength was set to 265 nm, slit width 2 nm and excitation data range between 300 nm and 530 nm. Time resolved studies were done using a time correlated single photon counting (TCSPC) system from Horiba Yovin (Model: Fluorocube-01-NL). Samples were excited at 376 nm using a picosecond diode laser (Model: Pico Brite-375L). All NMR characterizations were carried out on a Bruker AV 400 MHz spectrometer. Compound concentrations were in the range 1–10 mmol in (CD3)2SO and CDCl3 and mass spectra were recorded using a Bruker micrOTOF-Q II positive mode electrospray ionization mass spectrometer.Acknowledgements
AKD is grateful to the Department of Science and Technology (grant no. SR/FT/CS-67/2010), New Delhi, India, for financial support. DBR and IM are indebted to CSIR, New Delhi, India, for research fellowships. We thank SAIF IIT Bombay for use of the SEM.References
- J. M. Lehn, Supramolecular chemistry : concept and perspective, VCH, Weinheim, 1995 Search PubMed.
- (a) S. Kiyonaka, K. Sada, I. Yoshimura, S. Shinkai, N. Kato and I. Hamachi, Nat. Mater., 2004, 3, 58 CrossRef CAS; (b) A. Wada, S. Tamaru, M. Ikeda and I. Hamachi, J. Am. Chem. Soc., 2009, 131, 5321 CrossRef CAS.
- (a) E. Genove, C. Shen, S. G. Zhang and C. E. Semino, Biomaterials, 2005, 26, 3341 CrossRef CAS; (b) J. P. Jung, A. K. Nagaraj, E. K. Fox, J. S. Rudra, J. M. Devgun and J. H. Collier, Biomaterials, 2009, 30, 2400 CrossRef CAS.
- S. Bhuniya, S. M. Park and B. H. Kim, Org. Lett., 2005, 7, 1741 CrossRef CAS.
- Z. M. Yang, G. L. Liang, M. L. Ma, A. S. Abbah, W. W. Lu and B. Xu, Chem. Commun., 2007, 843 RSC.
- Z. M. Yang, H. W. Gu, D. G. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440 CrossRef CAS.
- G. John, G. Zhu, J. Li and J. S. Dordick, Angew. Chem., Int. Ed., 2006, 45, 4772 CrossRef CAS.
- B. G. Xing, C. W. Yu, K. H. Chow, P. L. Ho, D. G. Fu and B. Xu, J. Am. Chem. Soc., 2002, 124, 14846 CrossRef CAS.
- (a) N. Amdursky, E. Gazit and G. Rosenman, Adv. Mater., 2010, 22, 2311 CrossRef CAS; (b) R. Orbach, L. Adler-Abramovich, S. Zigerson, I. Mironi-Harpaz, D. Seliktar and E. Gazit, Biomacromolecules, 2009, 10, 2646 CrossRef CAS; (c) J.-B. Guilbaud, E. Vey, S. Boothroyd, A. M. Smith, R.V. Ulijn, A. Saiani and A. F. Miller, Langmuir, 2010, 26, 11297 CrossRef CAS; (d) R. P. Nagarkar, R. A. Hule, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2008, 130, 4466 CrossRef CAS; (e) E. F. Banwell, E. S. Abelardo, D. J. Adams, M. A. Birchall, A. Corrigan, A. M. Donald, M. Kirkland, L. C. Serpell, M. F. Butler and D. N. Woolfson, Nat. Mater., 2009, 8, 596 CrossRef CAS.
- (a) A. L Boyle and D. N. Woolfson, Chem. Soc. Rev., 2011, 40, 4295 RSC; (b) E. Gazit, Chem. Soc. Rev., 2007, 36, 1263 RSC.
- A. R. Hirst, S. Roy, M. Arora, A. K. Das, N. Hodson, P. Murray, S. Marshall, N. Javid, J. Sefcik, J. Boekhoven, J. H. van Esch, S. Santabarbara, N.T. Hunt and R. V. Ulijn, Nat. Chem., 2010, 2, 1089 CrossRef CAS.
- Z. Yang, K. Xu, Z. Guo, Z. Guo and B. Xu, Adv. Mater., 2007, 19, 3152 CrossRef CAS.
- (a) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652 CrossRef CAS; (b) J. Boekhoven, A. M. Brizard, K. N. K. Kowlgi, G. J. M. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2010, 49, 4825 CrossRef CAS.
- R. T. S. Lam, A. Belenguer, S. L. Roberts, C. Naumann, T. Jarrosson, S. Otto and J. K. M. Sanders, Science, 2005, 308, 667 CrossRef CAS.
- N. Sreenivasachary and J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5938 CrossRef CAS.
- A. K. Das, A. R. Hirst and R. V. Ulijn, Faraday Discuss., 2009, 143, 293 RSC.
- F. H. Carpenter, J. Am. Chem. Soc., 1960, 82, 1111 CrossRef CAS.
- Y. Chen and G. Liang, Theranostics, 2012, 139 CrossRef.
- M. A. Greenfield, J. R. Hoffman, M. O. de la Cruz and S. I. Stupp, Langmuir, 2010, 26, 3641 CrossRef CAS.
- S. Mukhopadhyay, U. Maitra, G. Krishnamoorthy, J. Schmidt and Y. Talmon, J. Am. Chem. Soc., 2004, 126, 15905 CrossRef CAS.
- I. Maity, D. B. Rasale and A. K. Das, Soft Matter, 2012, 8, 5301 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Decay parameters, fluorescence spectra, % of conversion by HPLC, rheology, experimental part and synthesis details. See DOI: 10.1039/c2ra21334f |
| This journal is © The Royal Society of Chemistry 2012 |