DOI:
10.1039/C2RA21291A
(Paper)
RSC Adv., 2012,
2, 12438-12446
Nitrogen-doped graphene stabilized gold nanoparticles for aerobic selective oxidation of benzylic alcohols†
Received 26th June 2012, Accepted 17th October 2012
First published on 19th October 2012
Abstract
Increasing efforts have been made to fabricate Au/graphene composites due to the fascinating properties of both graphene and gold. Some Au nanoparticles with an average size of tens of nanometers were directly deposited on reduced graphene oxide (RGO), utilizing the residual oxy-functional groups as the “hitching post” of the nanoparticles. Some functional groups, such as amino and thiol, were attached to the surface of the graphene in order to stabilize gold nanoparticles with a smaller particle size (<5 nm in general). Unfortunately, most of these strategies result in Au particles with limited exposed atoms, which is certainly a disadvantage for their application, such as catalysis. Introduction of nitrogen heteroatoms into the framework of graphene can not only modulate the electronic structure, but also change the surface properties of the graphene. In this work, naked Au nanoparticles with an average size of about 2–4 nm were fabricated on nitrogen-doped graphene nanosheets (NG) via the direct simple reduction method. The Au/NG nanocomposites were characterized by XRD, XPS, TEM, AFM and Raman. It was revealed that the nitrogen atoms doped in NG, rather than defects or oxygen moieties, play an essential role in stabilizing Au NPs. It was also found that the initial reaction rate of benzyl alcohol oxidation over the Au/NG catalyst is about ten fold higher than that over Au/graphene catalysts. Our findings may provide a clue of nitrogen incorporation to stabilize uncapped noble metal nanoparticles on graphene or other inorganic oxide supports, such as TiO2.
Introduction
Catalysis by gold has currently become a hot topic of much current interest in chemistry, since Haruta and Hutchings found that nanoparticulate Au is an effective catalyst for the low-temperature CO oxidation and the hydrochlorination of ethyne in 1980s.1,2 Several recent reviews have comprehensively highlighted the importance of the research in gold catalysis, especially gold-catalyzed organic transformation reactions.3–9 Of particular interest to us is the selective oxidation of alcohols, which is one of the most important reactions in chemical industry.10–12 The ideal “green” processes for the reaction should be carried out at atmospheric pressure and room temperature, in aqueous media or under solvent-free conditions, using air or molecular oxygen as oxidant. Gold is well-established to be a highly desirable catalyst for the aerobic selective oxidation of alcohols to the corresponding aldehydes, ketones, and carboxylic acids in aqueous media because of its chemical stability to oxygen,13,14 whereas the heterogenous catalytic processes highly depend on the shape and size of gold.15 Among all parameters affecting gold activity and stability, the role of the support appears to be more and more important,16 because it can modulate the size, shape, and surface electronic structure of gold.Most reports on supported Au catalysts commonly involve inorganic metal oxide materials, including TiO2, Fe2O3, Al2O3, SiO2, and CeO2,17 but there is increasing interest in the use of carbon materials, including active carbon and carbon nanotubes, as supports because they have a number of advantages over oxide supports, such as higher chemcial stability, larger surface area, and lower cost.18 Recently, graphene, a single-atom layer of graphite possessing a unique two-dimensional structure, has received tremendous attention. This smart sp2-carbon packed monolayer material has brought more surprises, such as the quantum Hall effect, high carrier mobility at room temperature (ca. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1),19 large theoretical specific surface area (2630 m2 g−1),20 good optical transparency (ca. 97.7%),21 high Young's modulus (ca. 1 TPa),22 and excellent thermal conductivity (3000–5000 W m−1 K−1)23 as compared to carbon nanotubes. Owing to these unique properties, this fascinating material was often used as a building block for fabricating composite functional materials. Many types of graphene-based composites, including metal/graphene, oxide/graphene, polymer/graphene, and carbon nanotubes/graphene have been reported and applied in broad areas from batteries,24–27 supercapacitors,28–30 fuel cells,31–33 photovoltaic devices34–36 and photocatalysis37–39 to biosensors.40,41 Especially for Au/graphene composites, a number of preparative protocols, such as direct chemical reduction, and photochemical42 and microwave43,44 assisted reductions, have been developed to construct Au/graphene nanohybrids. However, the chemical inertness of graphene affects to a large extent the stabilization of gold nanoparticles and the achievement of a uniform distribution. To deal with the issue, chemical functionalization of the graphene surface have been suggested by many research groups. Oxy-functional groups are frequently introduced to act as nucleation sites and facilitate seeding and growth of Au nanoparticles, mainly based on the electrostatic interaction, steric blocking, and polymeric coordination. A research carried out by Muszynski et al. showed that Au nanoparticles (NPs) can be readily loaded on octadecyamine (ODA) fuctionalized graphene via chemical reduction of AuCl4− in THF.45 Chen and coworkers46 reported a wet-chemistry strategy to efficiently and uniformly decorate reduced graphene oxide with Au nanodots by in situ nucleation and growth of gold on the perylene thiol derivative (ETPTCDI) modified graphene surface. Hong et al.47 demonstrated that positively charged gold NPs with diameters of 2–6 nm were self-assembled on the surface of 1-pyrene butyric acid functionalized graphene nanosheets. Li et al.48 used 3,4,9,10-perylene tetracarbonxylic acid (PTCA) functionalized graphene as a scaffold for the nucleation and growth of Au NPs, giving a dense coverage of Au. However, for the polymer- and surfactant-stabilized Au nanoparticles, an unavoidable shortcoming is that polymeric coordination or steric blocking of the surface atoms often compromises the catalytic activity.44
000 cm2 V−1 s−1),19 large theoretical specific surface area (2630 m2 g−1),20 good optical transparency (ca. 97.7%),21 high Young's modulus (ca. 1 TPa),22 and excellent thermal conductivity (3000–5000 W m−1 K−1)23 as compared to carbon nanotubes. Owing to these unique properties, this fascinating material was often used as a building block for fabricating composite functional materials. Many types of graphene-based composites, including metal/graphene, oxide/graphene, polymer/graphene, and carbon nanotubes/graphene have been reported and applied in broad areas from batteries,24–27 supercapacitors,28–30 fuel cells,31–33 photovoltaic devices34–36 and photocatalysis37–39 to biosensors.40,41 Especially for Au/graphene composites, a number of preparative protocols, such as direct chemical reduction, and photochemical42 and microwave43,44 assisted reductions, have been developed to construct Au/graphene nanohybrids. However, the chemical inertness of graphene affects to a large extent the stabilization of gold nanoparticles and the achievement of a uniform distribution. To deal with the issue, chemical functionalization of the graphene surface have been suggested by many research groups. Oxy-functional groups are frequently introduced to act as nucleation sites and facilitate seeding and growth of Au nanoparticles, mainly based on the electrostatic interaction, steric blocking, and polymeric coordination. A research carried out by Muszynski et al. showed that Au nanoparticles (NPs) can be readily loaded on octadecyamine (ODA) fuctionalized graphene via chemical reduction of AuCl4− in THF.45 Chen and coworkers46 reported a wet-chemistry strategy to efficiently and uniformly decorate reduced graphene oxide with Au nanodots by in situ nucleation and growth of gold on the perylene thiol derivative (ETPTCDI) modified graphene surface. Hong et al.47 demonstrated that positively charged gold NPs with diameters of 2–6 nm were self-assembled on the surface of 1-pyrene butyric acid functionalized graphene nanosheets. Li et al.48 used 3,4,9,10-perylene tetracarbonxylic acid (PTCA) functionalized graphene as a scaffold for the nucleation and growth of Au NPs, giving a dense coverage of Au. However, for the polymer- and surfactant-stabilized Au nanoparticles, an unavoidable shortcoming is that polymeric coordination or steric blocking of the surface atoms often compromises the catalytic activity.44
Theoretical calculation showed that doping nitrogen in the lattice of graphene can tailor the electron configuration and surface chemical reactivity.49 Indeed, nitrogen doped graphene (NG) was found to be a potential material for oxygen reduction reaction (ORR),50–52 lithium ion batteries,21 electrochemical biosensors53 and so forth. Our previous study also demonstrated that nitrogen-doped graphene nanosheets represent a new chemical function of efficiently catalyzing aerobic alcohol oxidation.54 The tremendous change in chemical reactivity of graphene after nitrogen-doping modification was closely related to graphitic sp2 nitrogen species. In the present work, we introduced nitrogen heteroatoms into graphene lattice to provide the active sites for nucleation and growth of Au nanoparticles. It was found that NG can serve as a more desirable scaffold for controlling the size of Au NPs as compared to graphene. The statistic results revealed that the average particle size of Au supported on NG is between 2 and 4 nm, and is much smaller than those supported on graphene. Furthermore, the catalytic properties of this uncapped Au loaded NG nanocomposites was evaluated by the aerobic selective oxidation of benzylic alcohols under the ambient conditions. It is found that the initial reaction rate of benzyl oxidation catalyzed by Au/NG composites is about 10 times that catalyzed by Au/graphene.
Experimental section
Chemical reagents used for the preparation of samples include graphite powder, NaCl, concentrated H2SO4, NaNO3, KMnO7, H2O2, HCl, HAuCl4·4H2O, NaOH and NaBH4. They were all purchased from Sinopharm Chemical Reagent Co., Ltd. All these chemicals are of analytical grade purity and were used without further purification. Ammonia gas, supplied by Fuzhou Xinhang Industrial Gases Co., Ltd, is of industrial grade purity (>95%).Synthesis of graphene and nitrogen-doped graphene nanosheets
Details of the method for support (graphene and NG) preparation were described in our previous work.50 For the support preparation, graphite oxide (GO) was firstly made via a modified Hummers' method.55 To produce nitrogen-doped graphene nanosheets, a heat-treatment procedure of the as-synthesized GO at 1173 K under ammonia flow (300 ml min−1) was applied. The details of this procedure were described as follows. GO samples were placed into a Al2O3 boat in the center of a tube furnace. After flowing of NH3 for 30 mins, the temperature of the furnace was raised to 573 K in 15 mins, and then directly heated up to 1173 K at a heating rate of 5 K min−1. After 10 h of calcination at the indicated temperature, the furnace was cooled down naturally to room temperature. In order to completely remove the ammonia adsorbed on the surface of the samples, a He stream flowed through the furnace for more than 30 mins after nitridation. For comparison, graphene nanosheets were synthesized by heating GO at 1173 K under flowing He.Synthesis of Au/G and Au/NG nanocomposites
A simple impregnation method followed by a reduction process was applied for Au loading. Generally, a certain amount of graphene or nitrogen-doped graphene was placed in a beaker, and then an appropriate volume of HAuCl4 solution (10 mg ml−1) was added. After sonication for 30 min, the mixture was dried at 393 K for 5 h. The resulting powder was reduced by 0.1 mol L−1 NaBH4 aqueous solution for 1 h, and then washed thoroughly with deionised water by vacuum filtration, and finally dried overnight at 333 K. By varying the amount of HAuCl4 solution, Au/G-x and Au/NG-x nanocomposites with different ratios of gold were prepared, where x is denoted as the loading content of gold.Catalyst characterizations
X-ray powder diffraction patterns were recorded on a Bruker D8 Advance X-ray diffractometer using Cu-Kα1 radiation (λ = 1.5406 Å), 40 kV, 40 mA with a scanning speed of 0.2° (2θ) min−1. Transmission electron microscopy (TEM) images were collected on a JEOL model JEM 2010 EX microscope at an accelerating voltage of 200 kV. The XPS spectra were carried out on an ESCALAB 250 XPS System with a monochromatized Al Ka X-ray source (15 kV 200 W 500 μm pass energy = 20 eV). The Raman spectra were measured using a laser with an excitation wavelength of 785 nm at room temperature on a Renishaw Raman microscope. Tapping-mode atomic force microscopy (AFM) measurements were performed on Nanoscope IIIA system. Samples for AFM imaging were prepared by depositing ethanol suspensions of different samples on a freshly cleaved mica surface.Catalytic activity testing
The oxidation reactions were carried out in a flask containing 80 ml deionized water, 1.0 mmol alcohol, 30 mg of catalyst, and a stir bar. Unless otherwise indicated, the aerobic oxidation reactions were conducted in an oil bath at 333 K in a static O2 atmosphere (1.0 atm). The reaction solution was sampled periodically and analyzed by liquid chromatography (Waters 2487, which was equipped with an Agilent Zorbax SB-C18 column). All products were quantified using the external standard method. Kinetic measurements were conducted at a temperature range of 308–333 K. The conversion of alcohol was controlled below 5.0% in order to collect data. The conversion of alcohols and the selectivity to their corresponding aldehydes and acids were calculated as follows:
| Conversion = (CAD + CA)/C0 × 100% |
| Selectivity = (CAD or CA)/(CAD + CA) × 100% |
where C0 is the initial concentration of alcohols, CAD and CA is the concentration of aldehydes and acids, respectively.Results and discussion
Nitrogen-doped graphene (NG) was prepared by a postnitridation of graphite oxide (GO) in a flowing NH3 atmosphere at a high temperature of 1173 K, as reported in our previous work.54 Three spots were covered in the single procedure: (i) the exfoliation of graphite oxide to graphene oxide nanosheets by thermal expansion; (ii) thermal reduction of graphene oxide nanosheets to graphene nanosheets; (iii) nitrogen doping into graphene lattice. Besides, this facile synthesis method provides us with NG in gram-scale quantities, which undoubtedly benefits the large-scale production of metal-NG composites. For comparison, graphene nanosheets (G) as a support of gold catalysts was prepared by reduction of GO in a He atmosphere while keeping other conditions unchanged. A direct chemical reduction procedure using NaBH4 as a reductant was applied to obtain Au/NG and Au/graphene nanohybrids. The obtained catalysts were characterized by transmission electron microscopy (TEM), X-ray powder diffraction (XRD), visible Raman spectroscopy, and atomic force microscopy (AFM).Synthesis and structural characterization of nitrogen-doped graphene/gold nanocomposites
Fig. 1 shows the XRD patterns of NG, graphene, and the as-synthesized Au/NG-x and the Au/G-x composites with different Au weight percentages. Bare graphene shows only a weak diffraction line at around 26.6° (d = 0.34 nm) corresponding to the (002) reflection of graphene sheets,56 while NG represents two weak peaks at ca. 16.1° (d = 0.55 nm) and 22.9° (d = 0.39 nm), besides the diffraction line at ca. 26.6°, as a result of nitrogen incorporation. The high-temperature doping not only causes the nonuniform expansion along the c-axis direction, but also makes the arrangement of graphene layers more ordered as shown by the marked increase in intensity of the characteristic diffraction peak at 26.6°. Upon gold loading at a low weight percentage of 0.5 wt% (Au/G-0.5), no diffraction peaks indexed to metallic gold are discriminated from the background due to the low coverage of Au in these composites. With increasing the Au weight ratio, the peaks at 2θ values of 38.2°, 44.6°, 64.7°, and 77.5° belonging, respectively, to the (111), (200), (220), and (311) crystal planes of metallic gold can be discerned and gradually increase in intensity, whereas the diffraction lines of the supports NG and graphene lack any changes. It indicates that the Au loading has no effect on the structure of the support materials. |
| | Fig. 1 XRD patterns of the Au/NG-x composites and NG (A). XRD patterns of the Au/G-x composites and bare graphene (B). | |
Fig. 2 shows the TEM images of a series of Au/G-x composites. All samples have morphology resembling a crumpled onion skin with “ink spots” on them. The crumpling of graphene layers, commonly observed in graphene, is attributed to defective structures formed in the thermal treatment process. The “ink spots” are believed to be Au based from their high contrast feature. This will be clearly proved by HRTEM results below. Au is highly dispersed on the surface of the graphene lattice in the form of nanoparticles. For the Au/G-0.5 sample, the average size of Au particles is ca. 30 nm (Fig. 2A and B). With the increase of Au percentage, Au formed bigger particles on graphene, and shows a wider distribution of particle size in the region 40–100 nm (Fig. 2D, F, and H). The EDX pattern of the Au/G-4.0 sample (the inset to Fig. 2G) shows C, O, and Au elements, and a copper signal originating from the copper grid holder used. The results indicate that the reduced graphene oxides (also denoted as graphene) are not a perfect support of Au catalysts prepared by the direct chemical reduction method.
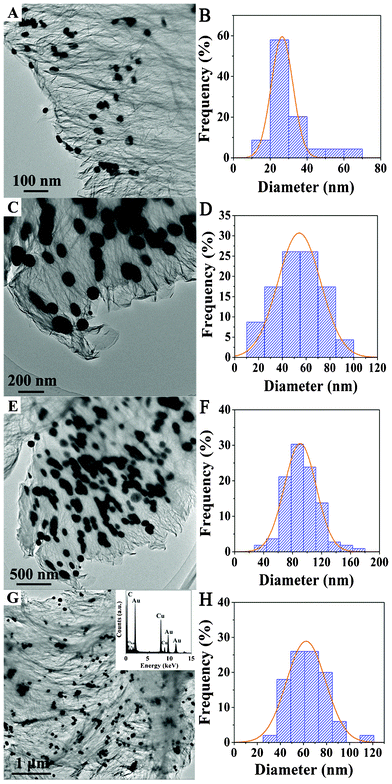 |
| | Fig. 2 TEM images and the size distribution of Au NPs for Au/G-0.5 (A and B), Au/G-1.5 (C and D), Au/G-3.0 (E and F) and Au/G-4.0 (G and H), respectively. The insertion in G is the EDX spectrum of Au/G-4.0. | |
Fig. 3 shows TEM images of a series of Au/NG-x samples. It appears that NG has the same morphology as graphene despite nitrogen incorporation. The EDX pattern (the inset to Fig. 3G) represents the N element in the Au/NG-4.0 sample in addition to C, O, and Au elements, indicating N is doped into the graphene lattice. Au particles are highly dispersed on the support surface. The HRTEM image (the inset to Fig. 3E) shows an interplanar spacing of about 0.24 nm corresponding to the (111) lattice plane of face-centered-cubic (fcc) gold. The average size of the gold nanoparticles is estimated to be ca. 3.6 nm as seen from the right histograms, far smaller than those supported on graphene with the same strategy (63 nm for the Au/G-x samples with more than 1.5 wt% of gold loading). More surprisingly, the average particle size of gold is unchanged with increasing the amount of loading in the studied region of 0.5–4.0 wt%. The significant difference in Au particle size between the Au/NG-x and Au/G-x samples implies that nitrogen doped in the graphene lattice plays a crucial role in stabilizing gold nanoparticles.
 |
| | Fig. 3 TEM images and the size distribution of Au NPs for Au/NG-0.5 (A and B), Au/NG-1.5 (C and D), Au/NG-3.0 (E and F) and Au/NG-4.0 (G and H), respectively. The insertion in E is the HRTEM image of Au NPs, and the insertion in G is the EDX spectrum of Au/NG-4.0. | |
To consolidate this conclusion, we further characterized the microstructure of the Au/NG-3.0 and Au/G-3.0 samples with atomic force microscopy (AFM). The obtained AFM results are in good agreement with the above TEM results, as shown in Fig. 4. The AFM image of the Au/G-3.0 sample (Fig. 4A) presents a large graphene nanosheet decorated with Au nanoparticles. The randomly chosen height profile demonstrates that the undoped graphene nanosheet has a thickness of ca. 2.9 nm and Au nanoparticles dispersed on the surface of graphene nanosheets has a height of ca. 60.4 nm. For the Au/NG-3.0 sample (Fig. 4B), a number of shining dots attributed to Au nanoparticles with a height of ca. 4.5 nm are discerned on the NG plane. The determined thickness of the NG nanosheets is between 2.7 and 3.1 nm (Fig. 4C), corresponding to a layer number of 8–9, which is consistent with that of graphene nanosheets and the previous results.54 One can thus conclude that the dominant contributor stabilizing gold nanoparticles is the nitrogen dopant, instead of the physical thickness of graphene nanosheets or the formed structural defects during the high-temperature exfoliation of GO. This is further verified by the equivalent ID/IG ratio, which is an indication of the number of structural defects and edge plane exposure,57 as shown in Raman spectra (Fig. S1 in ESI†).
 |
| | Fig. 4 AFM images (left) and height profiles (right) of Au/G-4.0 (A) and Au/NG-4.0 (B and C). | |
XPS characterization of nitrogen-doped graphene-gold nanocomposites
Previous reports suggested that the presence of oxygen functionalities or structural defects at the graphene surface provide reactive sites for the nucleation and growth of gold nanoparticles.58–61 To explore the nucleation, growth, and stabilization mechanism of these highly-dispersed gold nanoparticles on the NG substrate, we firstly carried out XPS characterization for the Au/NG-3.0 and Au/G-3.0 samples together with the bare NG. The obtained results are shown in Fig. 5. The NG substrate shows a broad N 1s signal in the high-resolution XPS spectrum. This signal could be fitted into three subpeaks centered at 398.0, 399.5, and 401.2 eV, indicating the occurrence of three kinds of nitrogen species in these NG nanosheets. According the literature,47 the subpeak at 398.0 eV corresponds to pyridinic N, and the one at 399.5 eV is ascribed to pyrrolic N. The binding energy of 401.2 eV should be attributed to graphitic N. For the Au/NG-3.0 sample, four main feature peaks, which are ascribed to C 1s, N 1s, O 1s and Au 4f core levels, are observed in the survey scan spectrum of the Au/NG-4.0 sample (Fig. S2 in ESI†). Interestingly, after Au loading, the N 1s XPS spectrum is significantly changed compared to the parent NG. The binding energy of nitrogen species shifts from 398.0 eV for pyridinic N, 399.5 eV for pyrrolic N and 401.2 eV graphitic N to 398.1, 400.5, and 402.5 eV, respectively. Moreover, it can be seen from the high-resolution XPS spectrum of Au that the Au 4f core level peaks at 87.5 eV due to Au4f5/2 and 83.8 eV due to Au4f7/2 are correspondingly shifted by 0.3 eV towards the low-energy direction compared to those (87.8 eV for Au4f5/2 and 84.1 eV for Au4f7/2) of the Au/G-3.0 sample. It indicates that there is a strong chemical interaction between the nitrogen dopants and Au nanoparticles. These Au nanoparticles act as electron acceptors while the nitrogen dopants play the role of electron donor. The large positive shifts for both pyrrolic N (Δ = 1.0 eV) and graphitic N (Δ = 1.3 eV) show that the strong Au–N interaction takes place primarily at the pyrrolic and graphitic N sites. It was estimated using photoelectron cross sections calculated by Scofield that the O atomic content is 2.0 at.% in NG, less than that (2.9 at%) in graphene; therefore, the contribution of the residual oxygen groups in NG can be completely excluded to the formation of highly-dispersed gold nanoparticles. |
| | Fig. 5 XPS analysis of Au/G (a), Au/NG (b), and NG (c) samples. | |
According to the above-reported results, it can be thus concluded that Au nanoparticles nucleated and grew at the pyrrolic and graphitic N sites. Some earlier studies have also shown that nitrogen can act as the anchoring site for platinum seeds.33,62 As such, these nitrogen heteroatoms in NG serve as the anchoring sites for Au seeds as shown by the strong Au–N interaction. We can thus give a clear picture of the nucleation and growth of gold nanoparticles on NG, as depicted in Scheme 1. Gold cations prefer to be first adsorbed at the N doping sites, and then are reduced to form a gold nucleus by the NaBH4 reductant. For the remaining AuCl4− ions, there are two routes to their destination: (i) adsorbed endlessly on the surface of as-formed gold nucleus and reduced to Au(0) by NaBH4, resulting in growth of the gold nucleus; (ii) adsorbed on a new reactive site and directly reduced to Au(0), consequently forming a new nucleus. The abundance of N anchoring sites on the flat plane of the NG facilitates the formation of more gold nuclei. It makes the large part of AuCl4− unavailable for the growth of Au seeds to nanoparticles by route (i) because of the Coulombic interaction between AuCl4− and graphitic N with a positive charge. This explanation is strongly supported by the fact that the average size of gold nanoparticles is not varied with increasing gold loading. Another role of nitrogen heteroatoms is as the stabilizers for Au nanoparticles. In the case of no nitrogen doping, these formed Au nuclei tend naturally to aggregate to form bigger particles due to the large surface energy. The double roles of nitrogen dopants make it come true to assemble well-dispersed gold nanoparticles on NG.
 |
| | Scheme 1 The proposed mechanism of gold formation on nitrogen-doped graphene. | |
Catalytic behavior of nitrogen-doped graphene–gold nanocomposites
It is well-known that gold catalysis strongly depends on its size. The smaller the size of the gold particles, the more active Au atoms are exposed. This will contribute a lot to the activity enhancement. In this work, we first evaluated the catalytic performance of these gold nanoparticles using the Au/NG-4.0 sample as a representative catalyst and the aerobic oxidation of primary and secondary alcohols in water as a model reaction. The obtained results are summarized in Table 1. For the aerobic oxidation of primary alcohols, we chose benzylic alcohols and 1-hexanol as representatives. The obtained oxidation products of benzylic alcohols are the corresponding aldehydes and carboxylic acids. The main oxidation product of benzyl alcohol (BA) is benzaldehyde in the case of a low conversion (6.3%) (Entry 6), but upon a high conversion (above 50%) benzylic acid is predominant (Entry 1). Compared to benzyl alcohol, these benzylic alcohols substituted by one electron-donating or -withdrawing group are more easily oxidized over these gold nanoparticles. Under the same reaction conditions, the conversion of p-fluorobenzyl alcohol and p-methoxybenzyl alcohol is 83% and 95%, respectively, significantly higher than that (67%) of benzyl alcohol. However, secondary alcohols are found to be relatively reluctant toward the aerobic oxidation, as listed in Entry 4. The Au/NG-4.0 catalyst exhibits a lower conversion (54%) for 1-phenylethanol compared to these primary benzylic alcohols, while a selectivity of 100% to the corresponding ketone is obtained. According to literature,63 the aerobic oxidation of benzylic alcohols is predominantly determined by a crucial step of α-H dehydrogenation, which causes the formation of a carbocation as the reactive intermediate or transition state. Primary benzylic alcohols contain two active α-H atoms, and thus are easily oxidized into carboxylic acids because of the thermodynamic permission for the oxidation of benzaldehyde by O2 to benzoic acid.64 The presence of the electron-donating CH3O– or electron-withdrawing F– group connected to the benzene ring facilitate the conversion, but they can suppress in minor part the second oxidation of benzaldehyde to benzoic acid. The selectivity to the corresponding carboxylic acid decreases in the order of benzyl alcohol (60%) > p-methoxybenzyl alcohol (54%) > p-fluorobenzyl alcohol (47%), and the selectivity to the corresponding aldehyde increases in reverse order. For the oxidation of 1-phenylethanol into acetophenone, the further oxidation doesn't take place because acetophenone doesn't contain the active α-H atom. One can thus conclude that the gold-catalyzed benzylic alcohol oxidation follows an α-H dehydrogenation pathway. The stability of the formed carbocation is a pivotal factor affecting the conversion of the reactant. This is further confirmed by the lower reactivity of 1-hexanol over these gold nanoparticles. As demonstrated in Entry 5, only 9.8% 1-hexanol are catalytically converted into hexanal under the indicated conditions. Oxidation of primary aliphatic alcohols to the corresponding aliphatic aldehydes remains a challenge in the aerobic oxidation of alcohols for most heterogeneous catalysts.
Table 1 Oxidation of various alcohols using Au/NG-4.0 with oxygen
| Entry | Alcohol | Time [h] | Conversion [%] | Selectivity [%] |
|---|
| Aldehyde/ketone | Acid |
|---|
| Reaction conditions: 0.1 mmol of alcohol dissolved in 80 ml H2O, 30 mg of Au/NG-4.0 catalyst, 1 atm O2, 343 K, 1300 rpm. Reaction conditions: 0.1 mmol of 1-hexanol dissolved in 5 ml acetonitrile, 30 mg of Au/NG-4.0 catalyst, 1 atm O2, 343 K, 1300 rpm. The reaction solution was analyzed by gas chromatography. Reaction conditions: 1 mmol of benzylic alcohol dissolved in 80 ml H2O, other conditions as a. |
|---|
| 1a | 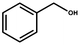 | 6 | 67 | 40 | 60 |
| 2a | 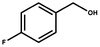 | 6 | 83 | 46 | 54 |
| 3a |  | 6 | 95 | 53 | 47 |
| 4a | 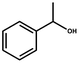 | 6 | 54 | 100 | — |
| 5b |  | 6 | 9.8 | 100 | — |
| 6c |  | 3 | 6.3 | 95 | 5 |
What advantages do these gold nanoparticles supported on NG have over the large gold nanoparticles loaded on graphene? To illustrate this issue, we performed a series of control experiments under the same reaction conditions. Table 2 shows the catalytic results of the selective aerobic oxidation of benzyl alcohol to benzaldehyde by O2 under the controlled conditions. We limited the conversion of benzyl alcohol below 5.0%, in order to accurately obtain some kinetic parameters. As can be seen clearly from the table, the conversion is low, but it still shows a clear increase with the weight percentage of gold for all catalysts. This is rational from the fact that the number of exposed Au atoms increases in such a case, which facilitates the benzyl alcohol transformation. However, a slight loss of selectivity to benzaldehyde is observed, accompanied by enhancement of the conversion. The further oxidation of benzaldehyde to benzoic acid is well-established to be the ringleader, which is inevitable for the benzyl alcohol oxidation with noble-metal catalysts. The obtained initial reaction rates (r0) are also listed also in Table 2 for all catalysts. We plotted r0 value vs. gold content and showed it in Fig. 6. The good linear correlation between r0 value and gold content further confirms that the reaction indeed occurs on gold active sites, instead of the support. It is noted that the intercepts of the two lines are zero. Technically speaking, the zero intercept implies that the contribution of the supports NG or graphene to the catalytic activity is completely negligible, but it is not the fact. It is inconsistent with our previous results that NG nanosheets can function as catalysts for the aerobic alcohol oxidation.50 A blank experiment using bare NG as a catalyst shows that r0 of the bare NG is equal to 0.08 mmol L−1 h−1 as shown by the asterisk in Fig. 6. It is significantly higher than that of Au/NG-0.1 and Au/NG-0.5. A coupling catalytic effect between gold and active nitrogen atoms doped in the graphene lattice doesn’t occur. This mainly originates from their strong interaction, as indicated by the above-reported XPS results. The Au–N interaction makes the catalytic function of NG disappear for catalyzing the BA oxidation, mainly because the active nitrogen sites on NG are occupied by gold nanoparticles.
 |
| | Fig. 6 The relationship between the initial reaction rate of BA oxidation over Au/NG-x and Au/G-x composites and the Au weight percentage. | |
Table 2 Comparison of catalytic activity between Au/NG-x and Au/G-x catalystsa
| Au content [wt%] | Au/NG-x | Au/G-x | r0(Au/NG-x)/r0(Au/G-x) |
|---|
| Conversion [%] | Selectivity [%] | r0 [mmol L−1 h−1] | Conversion [%] | Selectivity [%] | r0 [mmol L−1 h−1] |
|---|
| Amount of catalyst: 30 mg; substrate: 1 mmol benzyl alcohol dissolved in 80 ml H2O; O2 pressure: 1.0 atm; stirring speed: 1300 rpm; reaction temperature: 333 K; reaction time: 6 h. |
|---|
| 0.5 | 1.1 | >99 | 0.057 | 0.16 | >99 | 0.0048 | 11.9 |
| 1.5 | 3.3 | >99 | 0.25 | 1.2 | >99 | 0.029 | 8.5 |
| 3.0 | 6.3 | 97 | 0.47 | 1.3 | 98 | 0.056 | 8.4 |
| 4.0 | 7.3 | 93 | 0.60 | 1.8 | 97 | 0.073 | 8.2 |
The r0 ratio of Au/NG-x to Au/G-x, denoted as r0(Au/NG-x)/r0(Au/G-x) in the last column of Table 2, further highlights the profit of nitrogen heteroatoms doped into the graphene framework. Remarkably, the initial reaction rate is enhanced about 10 folds. It is interesting to note that the ratio drops from about 11.9 to 8.5 upon increasing the gold weight percentage from 0.5 wt% to 1.0 wt%, and keeps constant upon further increasing the gold weight percentage.
What is the nature of the activity enhancement? To clarify this issue, we carried out further kinetic analysis for the aerobic oxidation of BA over Au/NG-2.0 and Au/G-2.0 catalysts. Fig. 7A and C, respectively, displays their time curves of benzaldehyde (BAD) concentration at different temperatures. It clearly appears that, at an indicated temperature, the concentration of the product, benzaldehyde, increases linearly with reaction time. No benzoic acid or other by-products besides benzaldehyde were detected using liquid chromatography at the same time. The reaction rate r (r = dCBAD/dt, where CBAD is the concentration of BAD) is constant. On the other side, the r value varies with the reaction temperature. According to the Arrhenius equation of ln k = ln A − Ea/RT, we plotted ln k versus 1/T, where k is the rate constant obtained by linearly fitting the conversion curve of benzyl alcohol within the initial 60 mins of reaction at an indicated temperature. The plot of ln k versus 1/T is satisfactorily linear, as shown in Fig. 7B and D. For the Au/NG-2.0 catalyst, the slope and intercept are −6026 and 7.83, respectively, from which the apparent activation energy (Ea) and pre-exponential factor (A) calculated by the Arrhenius equation are 50.1 ± 4.0 kJ mol−1 and 2.51 × 103 mol−1 dm3 s−1, respectively. For the Au/G-2.0 catalyst, the slope and intercept are −5930 and 2.1, respectively. It can be calculated that the Ea and A values are equal to 49.3 ± 5.0 kJ mol−1 and 8.2 mol−1 dm3 s−1, respectively. Their activation energy (Ea) is equivalent and very close to that (51.4 kJ mol−1) reported for Ru/Al2O3 catalyzed aerobic benzyl alcohol oxidation using PhCF3 as solvent,65 but lower than that of NG (56.1 kJ mol−1).54 It suggests that the aerobic alcohol oxidation prefers to proceed on gold. Moreover, the similarity between the activation energy of gold supported on graphene and NG indicates that the surface gold atoms in either the Au/NG or Au/graphene composite have the same catalytic reactivity. This can be further confirmed by the fact that, for samples with Au at 0.5wt%, the ratio between TOF of Au/NG composite and Au/G composite is 1.8, approximating to 1.0 (see supporting information† for the details of the calculation). It is important to note that there is a drastic difference in pre-exponential factor (A) of Au/NG-2.0 and Au/G-2.0, which can well explain the above-reported fact that the Au/NG-x catalysts show a higher catalytic reactivity for the aerobic oxidation of benzylic alcohols than the Au/G-x catalysts.
 |
| | Fig. 7 Time curves of benzaldehyde concentration at different temperatures. Catalyst: 30 mg Au/NG-2.0 (A) or Au/G-2.0 (C); substrate: 80 ml 12.0 mmol L−1 aqueous solution of benzyl alcohol; O2 pressure: 1.0 atm; stirring speed: 1300 rpm. The corresponding Arrhenius plot of the benzyl alcohol oxidation with molecular oxygen (B for Au/NG-2.0 and D for Au/G-2.0). | |
Conclusions
In summary, highly dispersed gold nanoparticles with an average particle size of 3.6 nm were successfully fabricated on NG by a facile reduction method. The XPS characterization results reveal that the strong interaction of gold with the pyrrolic and graphitic N species doped into the graphene lattice play a pivotal role in stabilizing these gold nanoparticles. The aerobic oxidation of benzylic alcohol was used as a model reaction to evaluate the catalytic activity of Au nanoparticles under the mild reaction conditions. Compared to the Au/G composites with large gold particles, the as-prepared Au/NG nanocomposites show a more excellent catalytic activity. The initial reaction rate is significantly enhanced about 10 fold by the simple introduction of nitrogen heteroatoms in graphene networks due to the more exposed gold atoms accessible for reactants. The overall activation energy of the gold-catalyzed BA oxidation is estimated to be about 50.0 kJ mol−1, independent of the particle size of gold.What's more, this work demonstrates a promising strategy to fabricate “uncapped” metal nanoparticles with enhanced density of accessible catalytic sites. Other widely-used noble metals such as Ag, Pt and Pd, can be hopefully anchored on graphene and maintain most of the exposed active sites by the metal–N interaction.
Acknowledgements
This work was financially supported by the NSFC (Grants No. 21003021 and 21173044), the Natural Science Foundation of Fujian Province of P. R. China (2010J05024), the Science and Technology Project of Education Office of Fujian Province of P. R. China (JA12017), and National Basic Research Program of China (973 Program, No. 2012CB722607). We also express our gratitude to Miss Danmei Pan, from Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, for her assistance with the AFM analysis.References
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 16, 405–408 CrossRef.
- G. J. Hutchings, J. Catal., 1985, 96, 292–295 CrossRef CAS.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- P. C. Della, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077–2095 RSC.
- S. Carrettin, M. C. Blanco, A. Corma1 and A. S. K. Hashmi, Adv. Synth. Catal., 2006, 348, 1283–1288 CrossRef CAS.
- A. S. K. Hashmi, C. Lothschütz, M. Ackermann, R. Doepp, S. Anantharaman, B. Marchetti, H. Bertagnolli and F. Rominger, Chem.–Eur. J., 2010, 16, 8012–8019 CAS.
- B. K. Min and C. M. Friend, Chem. Rev., 2007, 107, 2709–2724 CrossRef CAS.
- A. Wittstock, V. Zielasek, J. Biener, C. M. Friend and M. Bäumer, Science, 2010, 327, 319–322 CrossRef CAS.
- G. -J. T. Brink, I. W. C. E. Arends and R. A. Sheldon, Science, 2000, 287, 1636–1639 CrossRef.
- R. A. Sheldon, I. W. C. E. Arends, G. -J. T. Brink and A. Dijksman, Acc. Chem. Res., 2002, 35, 774–781 CrossRef CAS.
- R. A. Sheldon, I. W. C. E. Arends and A. Dijksman, Catal. Today, 2000, 57, 157–166 CrossRef CAS.
- G. J. Hutchings, M. Brust and H. Schmidbaur, Chem. Soc. Rev., 2008, 37, 1759–1765 RSC.
- C. D. Pina, E. Falletta and M. Rossi, Chem. Soc. Rev., 2012, 41, 350–369 RSC.
- T. K. Sau, A. Pal and T. Pal, J. Phys. Chem. B, 2001, 105, 9266–9272 CrossRef CAS.
- M. Comotti, W. C. Li, B. Spliethoff and F. Schüth, J. Am. Chem. Soc., 2006, 128, 917–924 CrossRef CAS.
- M. Haruta, CATTECH, 2002, 6, 102–115 CrossRef CAS.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 RSC.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS.
- R. R. Nair, P. Blake, A. N. Grigorenko, K. S. Novoselov, T. J. Booth, T. Stauber, N. M. R. Peres and A. K. Geim, Science, 2008, 320, 1308 CrossRef CAS.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS.
- J. Zhu, T. Zhu, X. Zhou, Y. Zhang, X. W. Lou, X. Chen, H. Zhang, H. H. Hng and Q. Yan, Nanoscale, 2011, 3, 1084–1089 RSC.
- Z.-S. Wu, W. Ren, L. Xu, F. Li and H.-M. Cheng, ACS Nano, 2011, 5, 5463–5471 CrossRef CAS.
- S. Yang, X. Feng, S. Ivanovici and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 8408–8411 CrossRef CAS.
- H. Wang, L.-F. Cui, Y. Yang, H. S. Casalongue, J. T. Robinson, Y. Liang, Y. Cui and H. Dai, J. Am. Chem. Soc., 2010, 132, 13978–13980 CrossRef CAS.
- Y. Zhang, H. Li, L. Pan, T. Lu and Z. Sun, J. Electroanal. Chem., 2009, 634, 68–71 CrossRef CAS.
- F. Li, J. Song, H. Yang, S. Gan, Q. Zhang, D. Han, A. Ivaska and L. Niu, Nanotechnology, 2009, 20, 455602 CrossRef.
- F. Kim, J. Luo, R. Cruz-Silva, L. J. Cote, K. Sohn and J. Huang, Adv. Funct. Mater., 2010, 20, 2867–2873 CrossRef CAS.
- Y. Li, L. Tang and J. Li, Electrochem. Commun., 2009, 11, 846–849 CrossRef CAS.
- L. Dong, R. R. S. Gari, Z. Li, M. M. Craig and S. Hou, Carbon, 2010, 48, 781–787 CrossRef CAS.
- R. Imran Jafri, N. Rajalakshmi and S. Ramaprabhu, J. Mater. Chem., 2010, 20, 7114–7117 RSC.
- L. Gomez De Arco, Y. Zhang, C. W. Schlenker, K. Ryu, M. E. Thompson and C. Zhou, ACS Nano, 2010, 4, 2865–2873 CrossRef CAS.
- X. Li, H. Zhu, K. Wang, A. Cao, J. Wei, C. Li, Y. Jia, Z. Li, X. Li and D. Wu, Adv. Mater., 2010, 22, 2743–2748 CrossRef CAS.
- J. Song, Z. Yin, Z. Yang, P. Amaladass, S. Wu, J. Ye, Y. Zhao, W.-Q. Deng, H. Zhang and X.-W. Liu, Chem.–Eur. J., 2011, 17, 10832–10837 CrossRef CAS.
- H. Liu, S. Ryu, Z. Chen, M. L. Steigerwald, C. Nuckolls and L. E. Brus, J. Am. Chem. Soc., 2009, 131, 17099–17101 CrossRef CAS.
- Y. Zhang, Z.-R. Tang, X. Fu and Y.-J. Xu, ACS Nano, 2010, 4, 7303–7314 CrossRef CAS.
- N. Zhang, Y. Zhang, X. Pan, X. Fu, S. Liu and Y.-J. Xu, J. Phys. Chem. C, 2011, 115, 23501–23511 CrossRef CAS.
- H. Chang, L. Tang, Y. Wang, J. Jiang and J. Li, Anal. Chem., 2010, 82, 2341–2346 CrossRef CAS.
- H. Wang, Q. Zhang, X. Chu, T. Chen, J. Ge and R. Yu, Angew. Chem., Int. Ed., 2011, 50, 7065–7069 CrossRef CAS.
- X. Huang, X. Zhou, S. Wu, Y. Wei, X. Qi, J. Zhang, F. Boey and H. Zhang, Small, 2010, 6, 513–516 CrossRef CAS.
- H. M. A. Hassan, V. Abdelsayed, A. E. R. S. Khder, K. M. AbouZeid, J. Terner, M. S. El-Shall, S. I. Al-Resayes and A. A. El-Azhary, J. Mater. Chem., 2009, 19, 3832–3837 RSC.
- K. Jasuja, J. Linn, S. Melton and V. Berry, J. Phys. Chem. Lett., 2010, 1, 1853–1860 Search PubMed.
- R. Muszynski, B. Seger and P. V. Kamat, J. Phys. Chem. C, 2008, 112, 5263–5266 CrossRef CAS.
- X. Yang, M. Xu, W. Qiu, X. Chen, M. Deng, J. Zhang, H. Iwai, E. Watanabe and H. Chen, J. Mater. Chem., 2011, 21, 8096–8103 RSC.
- W. Hong, H. Bai, Y. Xu, Z. Yao, Z. Gu and G. Shi, J. Phys. Chem. C, 2010, 114, 1822–1826 CrossRef CAS.
- F. Li, H. Yang, C. Shan, Q. Zhang, D. Han, A. Ivaska and L. Niu, J. Mater. Chem., 2009, 19, 4022–4025 RSC.
- L. S. Panchakarla, K. S. Subrahmanyam, S. K. Saha, A. Govindaraj, H. R. Krishnamurthy, U. V. Waghmare and C. N. R. Rao, Adv. Mater., 2009, 21, 4726–4730 CAS.
- D. Deng, X. Pan, L. Yu, Y. Cui, Y. Jiang, J. Qi, W.-X. Li, Q. Fu, X. Ma, Q. Xue, G. Sun and X. Bao, Chem. Mater., 2011, 23, 1188–1193 CrossRef CAS.
- L. Feng, Y. Chen and L. Chen, ACS Nano, 2011, 5, 9611–9618 CrossRef CAS.
- Z. Luo, S. Lim, Z. Tian, J. Shang, L. Lai, B. MacDonald, C. Fu, Z. Shen, T. Yu and J. Lin, J. Mater. Chem., 2011, 21, 8038–8044 RSC.
- Y. Wang, Y. Shao, D. W. Matson, J. Li and Y. Lin, ACS Nano, 2010, 4, 1790–1798 CrossRef CAS.
- J. Long, X. Xie, J. Xu, Q. Gu, L. Chen and X. Wang, ACS Catal., 2012, 2, 622–631 Search PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- H. Guo, X. Wang, Q. Qian, F. Wang and X. Xia, ACS Nano, 2009, 3, 2653–2659 CrossRef CAS.
- D. Luo, G. Zhang, J. Liu and X. Sun, J. Phys. Chem. C, 2011, 115, 11327–11335 CAS.
- M. Büttner and P. Oelhafen, Surf. Sci., 2006, 600, 1170–1177 CrossRef.
- I. Suarez-Martinez, C. Bittencourt, X. Ke, A. Felten, J. J. Pireaux, J. Ghijsen, W. Drube, G. Van Tendeloo and C. P. Ewels, Carbon, 2009, 47, 1549–1554 Search PubMed.
- W. T. Wallace, B. K. Min and D. W. Goodman, J. Mol. Catal. A: Chem., 2005, 228, 3–10 CrossRef CAS.
- G. Goncalves, P. A. A. P. Marques, C. M. Granadeiro, H. I. S. Nogueira, M. K. Singh and J. Graácio, Chem. Mater., 2009, 21, 4796–4802 CrossRef CAS.
- G. Wu, D. Li, C. Dai, D. Wang and N. Li, Langmuir, 2008, 24, 3566–3575 CrossRef CAS.
- C. Shang and Z.-P. Liu, J. Am. Chem. Soc., 2011, 133, 9938–9947 CrossRef CAS.
- C. Marsden, E. Taarning, D. Hansen, L. Johansen, S. K. Klitgaard, K. Egeblad and C. H. Christensen, Green Chem., 2008, 10, 168–170 RSC.
- K. Yamaguchi and N. Mizuno, Chem.–Eur. J., 2003, 9, 4 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: Analysis of the Raman spectra, XPS survey scan of Au/NG-4.0 sample. See DOI: 10.1039/c2ra21291a |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.