Novel quadruple fluorescence of donor–acceptor benzoylthiourea derivatives
Wen
Yang
,
Wei
Zhu
,
Weiqun
Zhou
*,
Huanhuan
Liu
,
Yunlong
Xu
and
Jianfen
Fan
College of Chemistry, Chemical Engineering and Materials Science, Soochow University, 199 Ren'ai Road, Suzhou, P. R. China 215123. E-mail: wqzhou@suda.edu.cn; yangwen@suda.edu.cn; Tel: 86-0512-65884827
First published on 22nd August 2012
Abstract
Three donor–acceptor benzoylthiourea derivatives were characterized structurally by FTIR and NMR methods. Electronic spectra were investigated by UV absorption and steady fluorescence methods. Quadruple fluorescence bands on the fluorescence spectra were observed and re-assigned. Differently from the previous assignment for benzanilide (BA), the fluorescence bands with emission maxima of 480–524 nm were attributed to ESIPT (excited state intermolecular proton transfer). The fluorescence bands in the region 420–460 nm were explained by the TICT (twisted intramolecular charge transfer) model. Two short-wavelength fluorescence emissions at about 300 and 360 nm were assigned to the S2 state of two rotating isomers characteristic of the nature of the local excited (LE) state. The proposed mechanism for the emission was quantitatively supported by MP2 and CASSCF calculations.
Introduction
The property of double fluorescence emissions of benzanilide in solution has attracted attention for several decades.1–14 The short-wavelength band with maximum emission at 345 nm was interpreted as the normal fluorescence band of benzanilide [S1(LE) → S0].5 Because of the large Stokes shift of the long-wavelength fluorescence band in the region of 480–520 nm, O'Connell et al. concluded that changes of the geometry in the excited state were responsible for this fluorescence band.6 Azumaya et al.7 interpreted the emission band as a twisted intramolecular charge transfer (TICT) fluorescence. Heldt and Kasha8–11 regarded the fluorescence band as an overlapping of proton transfer (PT) fluorescence F2 [S1′(PT) → S0′(PT)] and TICT fluorescence F3 [S1′′(TICT) → S0(FC)]. Afterward, the TICT model was developed by Grabowski et al.12 The advanced model by Zachariasse et al.13,14 considered the intramolecular charge transfer state (ICT), which involved an amino nitrogen inversion by a solvent-induced two-level coupling, resulting in a change in the configuration of the amino nitrogen group from pyramidal in the ground state to planar in the excited state.Thiocarbonyl compounds have been of special interest to photochemists, and a substantial amount of data on their photophysical and photochemical properties has been reported.15–22 An important feature of the electronic spectroscopic properties of thiocarbonyl compounds is that the absorption bands corresponding to the S1 → S0, S2 → S0 and even higher electronic transitions are well resolved.15,23 Because of the large energy gap (typically in the range 5000–15000 cm−1) between the S2 and S1 state, strong S2 fluorescence (much stronger than S1 fluorescence) and weak S1 fluorescence is a common and regular feature in this class of compounds.24 Benzoylthiourea derivatives have structural characteristics of both benzanilide and thiocarbonyl compounds and these compounds exhibit interesting luminescent properties with a very strong influence from the molecular structure.25–32 A recent extensive investigation into the recognition and sensing of anions has been conducted.29–36 We have attributed the double fluorescence bands of FBMPT37 to the LE and CT fluorescence. However, the fluorescence mechanism is, as yet, not explicit. Which does the CT fluorescence belong to, ESIPT, TICT or another? And which excited state emits the local fluorescence? In this paper, three typical donor–acceptor thioureas have been investigated by photophysical characterization. With the aid of quantum chemical calculations, we attempt to explore clearly the luminescence mechanism to provide guidance on research for new fluorescent materials and their applications.
Experimental and theoretical method
The structures of N-benzoyl-N′-4-N,N-dimethylamidophenylthiourea (BDAPT) and N-(morpholinothiocarbonyl)-4-fluorobenzamide (MTFB) have been reported in the literature,38–40 but their fluorescent characteristics have not been reported. In this work, a new compound N-4-cyanobenzoyl-N′-4-N,N-dimethylamidophenylthiourea (CBDAPT) was synthesized, and in order to have an intercomparison of the fluorescent features, we also synthesized the aforementioned two compounds. The synthesis of CBDAPT was according to ref. 37. IR spectra were obtained in KBr discs using a Nicolet 170SX FT-IR spectrometer. NMR spectra were recorded in CDCl3 as the solvent, at room temperature and at 400 MHz on an INOVA 400 instrument. The absorption spectra were determined on a CARY50 UV-VIS spectrophotometer. Fluorescence measurements were carried out on a FLS920 analytical instrument. Quinine sulfate in 0.05 mol L−1 sulfuric acid is taken as the standard to detect the fluorescence quantum yield (QY).The ground-state minima structures were obtained by the MP241 method. The harmonic vibration frequencies were calculated to confirm the stability of the structures. Potential energy scans were carried out at a dihedral angle D around the C–N axis of benzamide. This angle was changed in steps of 10°. For each point, MP2 calculation with a 6-31G(d) basis set was performed. Subsequently, all of studied structures of the first excited state were optimized by the complete active space self-consistent field (CASSCF) method42,43 with an active space consisting of ten electrons in ten orbitals with the same basis set. The CASPT2 calculations44,45 were based on the structures of the first excited state. During the CASPT2 calculations, 65 orbitals have been kept frozen to make the calculation numerically feasible. The results were, however, not particularly sensitive to the exact number of orbitals kept frozen. The calculations were essentially stable with respect to change in active size and the number of states over which we averaged the energy during the orbital optimization. All of the calculations have been performed with the MOLPRO electronic code package.46
Results and discussion
The structure characteristics
Numbering on the studied benzoylthioureas is shown in Fig. 1. The data of elemental analysis, 1H NMR and FTIR of BDAPT and MTFB match well with the literature.38–40 | ||
| Fig. 1 Numbering of the studied benzoylthioureas. | ||
CBDAPT: orange solid, yield: 80%. Mp 181.4 °C. Elemental analysis: found: N 13.94, C 63.63, H 5.73; calc.: N 14.05, C 64.21, H 5.69%. 1H NMR (CDCl3): δ 2.984 (s, 6H, N(CH3)2), 6.717 (d, J = 8.8 Hz, 2H, C11–H, C13–H), 7.466 (d, J = 8.8 Hz, 2H, C10–H, C14–H), 7.805 (d, J = 8.4 Hz, 2H, C2–H, C6–H), 7.989 (d, J = 8.4 Hz, 2H, C3–H, C5–H), 9.288 (br s, 1H, N2–H2) ppm. FTIR (Nujol): ν/cm−1: 3456 (σO–H), 3223 (σN1–H1), 3138 (σN2–H2), 3032 (σPh–H), 2957 (σCH3, as), 2894 (σCH3, s), 2845 (σCH2, s), 2345 (σS–H), 2229 (νC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1670 (νC
N), 1670 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1610 (νPh), 1589 (νCC), 1522 (νN–C–N, as), 1444 (νCN), 1404 (νCN), 1346 (νPhN), 1264 (νC–S), 1233(νCN), 1186 (νCN), 1166 (δCCH), 1152 (δCCH), 1114 (νCC), 1072 (νCN), 1015 (νCN), 943 (γCH), 857 (τC–N), 818 (γCH), 781 (γCH), 748 (νCS), 687.9 (γCH).
O), 1610 (νPh), 1589 (νCC), 1522 (νN–C–N, as), 1444 (νCN), 1404 (νCN), 1346 (νPhN), 1264 (νC–S), 1233(νCN), 1186 (νCN), 1166 (δCCH), 1152 (δCCH), 1114 (νCC), 1072 (νCN), 1015 (νCN), 943 (γCH), 857 (τC–N), 818 (γCH), 781 (γCH), 748 (νCS), 687.9 (γCH).
Absorption spectra
We compare and critically analyze the characteristics of the absorption and fluorescence spectra in four different kinds of solvents: cyclohexane (a nonpolar aprotic solvent, ε = 2.023), THF (a weak polar aprotic solvent, ε = 7.58), acetonitrile (a strong polar aprotic solvent, ε = 36.64), and methanol (a strong polar protic solvent, ε = 32.63). The absorption spectra in the studied solvents are shown in Fig. 2. The maximum absorption wavelengths λa are listed in Table 1. The normalized steady fluorescence spectra are displayed in Fig. 3. The excitation wavelengths are displayed in the figures. The maximum emission wavelengths λmax of the fluorescence bands in the studied solvents are shown in Table 2. | ||
| Fig. 2 Absorption spectra of the studied compounds in different organic solvents. | ||
 | ||
| Fig. 3 Steady fluorescence spectra of three benzoylthioureas in the studied solvents. | ||
| λ a | Compound | Cyclohexane | THF | Acetonitrile | Methanol |
|---|---|---|---|---|---|
| λ a1 | BDAPT | 237 | 239 | 245 | 244 |
| CBDAPT | 246 | 246 | 247 | 242 | |
| MTFB | 250 | 238 | 238 | 238 | |
| λ a2 | BDAPT | 283 | 281 | 275 | 274 |
| CBDAPT | 304 | 289 | 287 | 283 | |
| MTFB | 292 | 285 | 282 | 282 | |
| λ a3 | BDAPT | 357 | 350 | 355 | 355 |
| CBDAPT | 370 | 370 | 370 | 370 | |
| MTFB | 350 | 350 | 350 | 350 |
| Cyclohexane | THF | Acetonitrile | Methanol | ||||||
|---|---|---|---|---|---|---|---|---|---|
| λ e | Compound | λ f/nm | QY/10−3 | λ f/nm | QY/10−3 | λ f/nm | QY/10−3 | λ f/nm | QY/10−3 |
| λ a1 | BDAPT | 306 | 12.4 | 358, 505 | 0.9, 3.6 | 300 | 3.6 | 298, 427 | 1.2, 0.6 |
| CBDAPT | 314 | 23.2 | 336, 488 | 4.3, 3.1 | 328 | 1.6 | 299, 355 | 0.22, 0.13 | |
| MTFB | 318, 408 | 5.6, 2.8 | 332, 428 | 2.1, 4.1 | 350, 454 | 3.6, 1.7 | — | — | |
| λ a2 | BDAPT | — | — | 358, 505 | 8, 8 | 358 | 1.8 | 422 | 2.0 |
| CBDAPT | — | — | 488 | 138.1 | — | — | — | — | |
The exhibited absorption bands of three compounds can be divided into three wavelength regions, which are similar to that of BA. The weak absorption bands in the region 350–370 nm have small molar absorptivities with values of 500–2000 L mol−1 cm−1. The absorption bands are very wide and the band half-width (BHW) reaches about 5000 cm−1. No significant change in the λa is observed in all studied solvents. Thus, we consider the weak and broad absorption bands as resulting from the charge transfer transition. The λa of the other two absorption bands are in the regions 274–304 and 237–250 nm, respectively. Because of the large molar absorptivities of 1 × 104 L mol−1 cm−1, these two bands are assigned to the π → π* transition. The absorption bands in the range 274–304 nm shift to slightly higher energy as the polarity of the solvents increases. This abnormal blue shift acts against the rule that a π → π* transition results in red shifting.
Steady fluorescence spectra
The fluorescence excitation spectra (Fig. 3) of these three compounds are similar to their UV absorption spectra. As with as other benzoylthioureas, the studied compounds exhibit faint fluorescence in the studied solvents at a concentration of 2 × 10−5 mol L−1. The absence of the mirror image relationship between the fluorescence and absorption spectra indicates clearly that a significant intramolecular relaxation has occurred upon photoexcitation. In the non-polar solvent cyclohexane, with an excitation wavelength of 245–260 nm, the λmax of the fluorescence bands are at 306 and 314 nm for BDAPT and CBDAPT, respectively. For MTFB, there is only one weak fluorescence band with λmax at 408 nm. Upon increasing the excitation wavelength, no other fluorescence bands can be detected. In the polar solvent, THF (ε = 7.58), when the same excitation wavelength is selected, double fluorescence bands are observed in all of the three compounds. The λmax of the high energy fluorescence bands are between 332 and 358 nm. The λmax of the low energy fluorescence bands increases from 428 to 505 nm in the sequence MTFB, BDAPT and CBDAPT. In the much more polar solvent acetonitrile, at an excitation wavelength of 245–260 nm, double fluorescence bands with λmax at 350 and 454 nm are detected for MTFB. However, with the same excitation wavelength, BDAPT and CBDAPT show single emission bands at 300 and 328 nm, respectively. In the strong polar protic solvent methanol, no fluorescence band is detected in MTFB, whereas BDAPT and CBDAPT exhibit double fluorescence bands. The λmax values of the high energy fluorescence bands are at about 300 nm, and the λmax of the low energy fluorescence bands are at 427 and 355 nm for BDAPT and CBDAPT. Upon changing the excitation wavelength, no significant change in the λmax of any emission bands is observed.As we can see, the fluorescence bands can be divided into four emission regions, corresponding to at least four different fluorescence states. The broad fluorescence bands of 480–524 nm, assigned by Kasha and co-workers,8–11 appeared in the fluorescence spectra of BA, MBA and FBMPT8–11,37 and are referred to as F4. The bands in the region 420–460 nm are regarded as F3. The other two emission bands with λmax at about 360 and 300 nm are denoted F2 and F1.
In general, three mechanisms may be considered as the origin of the multifluorescence bands. (a) The compound exists in different conformations in the ground and excited states. These conformations have different energies and are separated by a significant energy barrier. The different emission rates are the results of equilibrating conformers with different fluorescence lifetimes. (b) Upon irradiation, different transitions occur and independent excited states come into being. The excited states are deactivated by fluorescence, however, with significantly different emission rates. (c) There are excited-state intramolecular charge-transfer (ICT) states. In the excited state, the ICT state may emit at different wavelengths and with different fluorescence lifetimes. Heldt and Kasha8–11 regarded the long-wavelength fluorescence band of BA as an overlapping of proton transfer (PT) fluorescence F2 [S1′(PT) → S0′(PT)] and TICT fluorescence F3 [S1′′(TICT) → S0(FC)]. The F4 band appears at λmax = 505 nm for BDAPT in THF, which is similar to that of BA. According to Kasha's suggestion, the F4 fluorescence bands should belong to TICT. However, the fluorescence band with λmax at 505 nm is absent in MTFB. In the molecular structure of MTFB, the active proton has disappeared due to the existence of the intramolecular non-bonding interaction N–H⋯F, so the intermolecular proton transfer should not occur. We can only consider the F4 fluorescence of BDAPT in THF as ESIPT fluorescence.47–51 ESIPT is phototautomerization in the excited state via an intramolecular hydrogen bond involving transfer of the proton to the electronegative atom. Significant intramolecular relaxation takes place upon photoexcitation and this relaxation should increase the Stokes shift. In the studied fluorescence spectra in THF, the Stokes shift of the fluorescence band of BDAPT reaches 256 nm. The BHW is 2940 cm−1 and this band is annihilated in more polar solvents, such as acetonitrile and methanol. All characteristics of the large Stokes shift, the large BHW and the annihilation in strong polar solvents refer the F4 band to ESIPT.
Both BDAPT and MTFB emit an F3 band, which appears at 420–460 nm and also have large Stokes shifts and a large BHW. For example, for BDAPT, upon excitation at 257 nm, the Stokes shift of F3 reaches 170 nm in methanol, while the largest BHW is 2834 cm−1 in THF for MTFB. Additionally, the bathochromic shifts of F3 are observed as the polarity of the solvents increases. The λmax red-shifts from 408 nm in cyclohexane to 428 nm in THF and to 454 nm in acetonitrile. As noted by Kasha and co-workers,8–11 the large Stokes shift of F3 is indicative of a large change in geometry prior to fluorescence. The solvent-induced shift in the fluorescence maxima supports the assignment of F3 to a TICT state; thus, we regard the F3 as a TICT state. For MTFB, the benzoyl is taken as an acceptor, and the N-morpholino is regarded as a donor, while for BDAPT, because of the addition of a strong donor, N,N-dimethylamido, the charge transfer should be much stronger. Introduction of the acceptor group, cyano, to the para-benzoyl in BDAPT, leading to CBDAPT, should further increase charge transfer. We were not been able to determine a separate F3 band in the fluorescence spectra of CBDAPT and it is possible that the spectra have been incorrectly assigned. However, we find an abnormally broad fluorescence band appearing in the region of F4 and the BHW in THF reaches 4536 cm−1. The introduction of the cyano group increases the intramolecular charge transfer, which leads to the hypsochromic shift of F4 and the hyperchromic effects of F3, and likely causes the superposition of F3 and F4. Therefore, for CBDAPT, we consider the fluorescence band with λmax = 488 nm in THF as a mixture of intramolecular charge transfer and ESIPT.
Upon excitation at 260 nm, the λmax of F1 and F2 appear at about 300 and 360 nm for all the studied compounds. The Stokes shifts are 50–100 nm. Both fluorescence bands show a red-shifted with the increase in polarity of the solvents. Taking MTFB as an example, the λmax of F1 shows a bathochromic shift from 318 nm in cyclohexane to 332 nm in THF. With regard to the moderate Stokes shift, at the suggestion of Kasha,8–11 we attribute both F1 and F2 to the local excited (LE) states.
Quantum chemical calculations
Quantum chemical calculations are carried out using BDAPT as an example. The potential energy curves depicted in Fig. 4 are based on the structure of the first excited state using the CASPT2 method. Two minimum energy configurations are shown in the curve of S0 at 146.26 and 37.91°. The latter is 0.42 eV higher in energy than the former. The maximum energy point is located at 90°, and the energy barrier is calculated to be 0.89 eV, whereas two peaks and one trough are exhibited in the potential energy curves of the other states, T1, S1 and S2. The maximum energy points of T1, S1 and S2 correspond to the minimum energy point of S0, and the minimum energy points of T1, S1 and S2 correspond to the maximum energy point of S0. | ||
| Fig. 4 Potential energy surfaces of the first singlet excited state structure for C8–N1 rotation in BDAPT calculated by the CASPT2 method. | ||
The energy gaps between the S1 and T1 states at the maximum points are very small due to the relatively large spin–orbit coupling owing to the modest “heavy-atom effect” of the S atom.52–55 The energy gaps between the S1 and T1 states at 146.26 and 37.91° are only 0.06 and 0.08 eV, respectively, which are close to those of other thiocarbonyl compounds.52,53 In other words, there are the conical intersections between S1 and T1. When the electrons are excited to the first excited single state, they prefer to deactivate in the form of intersystem crossing (IC). The largest energy gap is 0.5 eV at 90°. It is difficult for IC to occur at this point. Because the probability of molecules being at a high energy level (at 90°) in the ground state is small, the excitation molecules at 90° deactivate in charge transfer and emit very weak S1 fluorescence. The energy gap of the charge transfer transition from the S1 state at 90° to the S0 state at 37.91° is 3.12 eV. The corresponding experimental energy of the λmax of F3 is 2.90 eV (427 nm) in methanol and the error between experiment and calculation is only 0.22 eV.
As we know, the charge-transfer transition must lead to a significant rearrangement of the electron density. For BDAPT, the frontier molecular orbitals (FMOs) of the minimum structure obtained by the MP2 method are shown in Fig. 5. The charge-transfer transition is characterized by electron density transfer from the toluidine ring to acylthiourea.
 | ||
| Fig. 5 Frontier molecular orbitals (FMOs) of BDAPT in the ground state by the MP2 method. | ||
Fig. 6 shows the optimized structures of the ground state using the MP2 method and the first excited state using the CASSCF methods for DBAPT. The torsion angle D(C1–C7–N1–C8) of the ground state is 21.5° and the molecule is nearly planar, whereas in the structure of the first excited state, the torsion angle D is 71.3° and the benzoyl and toluidine parts are nearly perpendicular. The molecular structure of DBAPT is extremely twisted in the first excited state. As noted by Kasha and co-workers,8 the large Stokes shift is indicative of a large change in geometry prior to fluorescence emission. The charge transfer transition from the S1 state to the S0 state should twist the dihedral angle D. Thus, the lowest energy transition of S1 → S0 is regarded as the TICT state, which emits fluorescence at λmax = 427 nm in methanol.
 | ||
| Fig. 6 Optimized structures of two tautomers of BDAPT (left: the ground state; right: the first excited singlet state). | ||
The strong S2 fluorescence is a common and regular feature of thiocarbonyl compounds.52,53 From Fig. 4, for DBAPT, the largest energy gap between S2 and S1 is 1.22 eV. Because of the typically large energy gap and the difference in symmetry characteristics between S2 and S1, we consider the local fluorescence as S2 fluorescence. The large energy barrier, corresponding to rotating the C8–N1 bond in the ground state, is 0.89 eV. We suggest that the two emissive species, F1 and F2, correspond to two excited-state conformations of the S2 fluorescence. Both conformations have different energy levels and are separated by a rotating barrier. For the two conformations of the minimum energy points corresponding to 37.91 and 146.26° (Fig. 4) of the ground state, the Franck–Condon transition energies of S0 → S2 are 4.62 and 4.04 eV, respectively. The corresponding experimental maximum emission energies of F1 in cyclohexane and F2 in THF are 4.05 eV (306 nm) and 3.46 eV (358 nm). The errors between the experiments and the calculation are close to 0.5 eV. As in the work done by Albrecht,56 the energy calculated for the transition is overestimated, which is a consequence of the approximation inherent to CASSCF. Because of the large energy gaps between S2 and S1 and the small energy gaps between T1 and S1, the lifetime of this S2 state should be longer than that of the common S2 state, and the molecule emits S2 fluorescence. Additionally, due to the small energy gap between S1 and T1 and the ultrafast intersystem crossing to the T1 state, FC fluorescence from the S1 state is insignificant.
Because of the existence of the active protons, the molecules of both BDAPT and CBDAPT can transfer the active protons in the form of intramolecular hydrogen bonds and produce thiol tautomers. FTIR spectra showing the stretching vibration frequencies at 2359.7 and 2345 cm−1 for BDAPT and CBDAPT, respectively, indicate the existence of the thiol tautomer. For BDAPT, both optimized structures of the thiol tautomer in the ground and first excited states using the MP2 and CASSCF methods are also displayed in Fig. 6. The calculated energy levels are shown in Table 3. The energy gap of S1T relative to S1K (S1T: the first excited state of the thiol tautomer, S1K: the first excited state of the keto tautomer) is 2.53 eV, corresponding to the experimental maximum emission energy, 2.46 eV (505 nm) in THF.
| Energy level | Relative energy/eV | |
|---|---|---|
| Keto-tautomer | Thiol-tautomer | |
| S0 | 0 | 1.48 |
| S1 | 2.56 | 5.09 |
| S2 | 4.62 | 7.67 |
| S3 | 5.65 | 7.8 |
Abnormal blue shifts of the absorption bands in the region 274–305 nm are observed, and the F1 bands of BDAPT and CBDAPT also show an exceptional blue shift in acetonitrile and methanol. The abnormal blue shift may be attributed to the intermolecular interactions between the thioureas and the solvent molecules. Being limited by computer resources, we only consider the molecular complexes of the short-range intermolecular hydrogen bond interactions of the first solvent-shell as the solvent effect. Taking BDAPT as an example, the optimized complexes with methanol and acetonitrile using the MP2 method are displayed in Fig. 7.
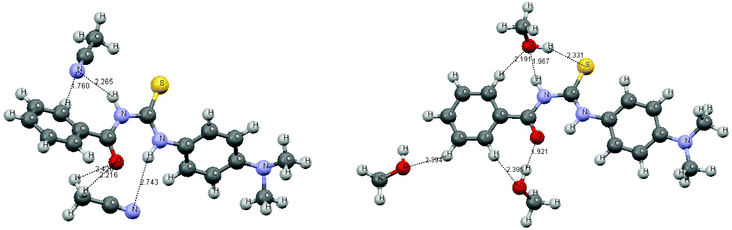
Conclusions
In comparison with BA, the fluorescence emission spectra are changed, and the fluorescence emission mechanism changes subsequently because of the insertion of the chromophore, thiocarbonyl. We regard the F4 bands in the region 480–524 nm as ESIPT fluorescence. This conclusion is made on the basis that (1) the F4 bands of MTFB disappear due to the absence of the active proton; (2) both Stokes shift and BHW are large; (3) F4 bands are annihilated in strong polar solvents; (4) FT-IR spectra exhibit the stretching vibration frequency –SH showing the existence of the thiol tautomer; and (5) the energy gap of S1T/S1K is close to the experimental maximum emission energy (error is only 0.07 eV). All of these indicate the characteristics of ESIPT.We consider the F3 bands in the region 420–460 nm as TICT. The fluorescence spectra characteristics, such as the solvent-induced shift in the emission maxima, the large BHW and the decrease of BHW with the increase in solvent polarity support the assignment of F3 to a TICT state. Moreover, the optimized structure of the first excited state is quite twisted. Additionally, the transition energy from S1 to S0 is close to the corresponding experimental emission energy of F3 for BDAPT in methanol. The error between the experiment and the calculation is only 0.22 eV.
Kasha has assigned the weaker fluorescence band (F1) of BA to a state with mixed n ← π* and π ← π* character and to a Franck–Condon singlet state or to a fluorescent impurity.8–11 This assignment does not confirm which state the Franck–Condon singlet state is, S1, S2 or S3. For the studied isomers, two high energy absorption bands have large molar absorptivities, therefore the corresponding short-wavelength fluorescence bands in the 300 and 360 nm regions should be assigned to the π ← π* transition with the Franck–Condon singlet state of the two rotating isomers. CASSCF calculations show the relatively small energy gaps of S1/T1 and the relatively large energy gaps of S2/S1, which cause the longer S2 lifetime of the state than that typically observed; further, the luminophores emit S2 fluorescence. Thus we attribute the two short-wavelength fluorescence bands to the S2 Franck–Condon singlet state of the two conformational isomers.
Acknowledgements
References
- S. Lucht and J. Stumpe, J. Lumin., 2000, 91, 203 CrossRef CAS.
- S. Lucht, J. Stumpe and M. Rutloh, J. Fluoresc., 1998, 8, 153–166 CrossRef CAS.
- D. L. Frederick and M. L. Timothy, J. Phys. Chem. A, 1998, 102, 5327–5332 CrossRef.
- D. L. Frederick and W. Liu, J. Phys. Chem. A, 1999, 103, 9678–9686 CrossRef.
- J. Heldt and M. Kasha, J. Mol. Liq., 1989, 41, 305–313 CrossRef CAS.
- E. J. O'Connell, M. Delmauro and J. Irwin, Photochem. Photobiol., 1971, 14, 189–195 CrossRef CAS.
- I. Azumaya, H. Kagechika, Y. Fujiwara, M. Itoh, K. Yamaguchi and K. Shudo, J. Am. Chem. Soc., 1991, 113, 2833–2838 CrossRef CAS.
- J. Heldt, D. Gormin and M. Kasha, Chem. Phys., 1989, 136, 321–334 CrossRef CAS.
- J. Heldt, D. Gormin and M. Kasha, Chem. Phys. Lett., 1988, 150, 433–436 CrossRef CAS.
- J. Heldt, D. Gormin and M. Kasha, J. Am. Chem. Soc., 1988, 110, 8255–8256 CrossRef CAS.
- G.-Q. Tang, J. Maclnnis and M. Kasha, J. Am. Chem. Soc., 1987, 109, 2531 CrossRef CAS.
- Z. R. Grabowski, K. Rotkiewicz, A. Siemiarczuk, D. J. Cowley and W. Baumann, Nouv. J. Chim., 1979, 3, 443 CAS.
- K. A. Zachariasse, M. Grobys, Th. von der Haar, A. Hebecker, Y. H'ichev and W. Kiihnle, J. Inf. Rec., 1996, 22, 553–560 CAS.
- Th. von der Haar, A. Hebecker, Y. H'ichev, Y.-B. Jiang, W. Kiihnle and K. A. Zachariasse, Recl. Trav. Chim. Pays-Bas, 2010, 114, 430–442 CrossRef.
- A. Maciejewski and R. P. Steer, Chem. Rev., 1993, 93, 67 CrossRef CAS.
- M. Szymanski, A. Maciejewski and R. P. Steer, J. Phys. Chem., 1988, 92, 2485 CrossRef CAS.
- M. Szymanski, A. Maciejewski and R. P. Steer, Chem. Phys., 1988, 124, 143 CrossRef CAS.
- G. Burdzinski, A. Maciejewski, G. Buntinx, O. Poizat, P. Toele, H. Zhang and M. Glasbeek, Chem. Phys. Lett., 2004, 393, 102 CrossRef CAS.
- G. Burdzinski, A. Maciejewski, G. Buntinx, O. Poizat and C. Lefumeux, Chem. Phys. Lett., 2004, 384, 332 CrossRef CAS.
- D. C. Moule and E. C. Lim, J. Phys. Chem. A, 2002, 106, 3072 CrossRef CAS.
- C. A. Emeis and L. J. Oosterhoff, J. Chem. Phys., 1971, 54, 4809 CrossRef CAS.
- J. P. Englebrecht, G. D. Anderson, R. E. Linder, G. Barth, E. Bunnenberg, C. Djerassi, L. Seamans and A. Moscowitz, Spectrochim. Acta, Part A, 1975, 31, 507 CrossRef.
- V. Ramamurthy and R. P. Steer, Acc. Chem. Res., 1988, 21, 300 CrossRef CAS.
- G. Burdzinski, A. Maciejewski, G. Buntinx, O. Poizat and C. Lefumeux, Chem. Phys. Lett., 2003, 368, 745 CrossRef CAS.
- W. Zhu, W. Yang, W. Q. Zhou, H. H. Liu, Sh. H. Wei and J. F. Fan, J. Mol. Struct., 2011, 1004, 74–81 CrossRef CAS.
- J. L. J. Blanco, P. Bootello, J. M. Benito, C. O. Mellet and J. M. G. Fernández, J. Org. Chem., 2006, 71, 5136–5143 CrossRef.
- F. Y. Wu, M. H. Hua, Y. M. Wua, X. F. Tana, Y. Q. Zhao and Z. J. Ji, Spectrochim. Acta, Part A, 2006, 65, 633–637 CrossRef.
- R. Martínez-Máñez and F. Sancenón, Coord. Chem. Rev., 2006, 250, 3081–3093 CrossRef.
- L. Nie, Z. Li, J. Han, X. Zhang, R. Yang, W. X. Liu, F. Y. Wu, J. W. Xie, Y. F. Zhao and Y. B. Jiang, J. Org. Chem., 2004, 69, 6449–6454 CrossRef.
- F.Y. Wu, L. H. Ma and Y. B. Jiang, Anal. Sci. (Suppl.), 2001, 17, 801–803 Search PubMed.
- F. Y. Wu, Z. Li, L. Guo, X. Wang, M. H. Lin, Y. F. Zhao and Y. B. Jiang, Org. Biomol. Chem., 2006, 4, 624–630 CAS.
- H. B. Li and H. J. Yan, J. Phys. Chem. C, 2009, 113, 7526–7530 CAS.
- W. X. Liu and Y. B. Jiang, J. Org. Chem., 2008, 73, 1124–1127 CrossRef CAS.
- X. Zhang, Ch. J. Wang, L. H. Liu and Y. B. Jiang, J. Phys. Chem. B, 2002, 106, 12432–12440 CrossRef CAS.
- R. Yang, W. X. Liu, H. Shen, H. H. Huang and Y. B. Jiang, J. Phys. Chem. B, 2008, 112, 5105–5110 CrossRef CAS.
- X. Zhang, X. Y. Sun, Ch. J. Wang and Y. B. Jiang, J. Phys. Chem. A, 2002, 106, 5577–5581 CrossRef CAS.
- W. Yang, W.-Q. Zhou and Z.-J. Zhang, J. Mol. Struct., 2007, 828, 46 CrossRef CAS.
- J. Imrich, T. Busova, P. Kristian and J. Dzara, Chem. Pap., 1994, 48, 42–6 CAS.
- C. R. Rasmussen, F. J. Villani, L. E. Weaner, B. E. Reynolds, A. R. Hood, L. R. Hecker, S. O. Nortey, A. Hanslin and M. J. Costanzo, Synthesis, 1988, 456–9 CrossRef CAS.
- H. Lothar, A. L. Ketty, A. C. Jorge, R. Rainer and B. Lothar, J. Fluorine Chem., 2009, 130, 453–460 CrossRef.
- A. El Azhary, G. Rauhut, P. Pulay and H. J. Werner, J. Chem. Phys., 1998, 108, 5185 CrossRef CAS.
- H. J. Werner and P. J. Knowles, J. Chem. Phys., 1985, 82, 5053 CrossRef CAS.
- P. J. Knowles and H. J. Werner, Chem. Phys. Lett., 1985, 115, 259 CrossRef CAS.
- H. J. Werner, Mol. Phys., 1996, 89, 645 CrossRef CAS.
- P. Celani and H. J. Werner, J. Chem. Phys., 2000, 112, 5546 CrossRef CAS.
- H. J.Werner, P. J.Knowles, R. Lindh, F. R. Manby, M. Schütz, P. Celani, T. Korona, A. G. Mitrushenkov, T. B. Rauhut, R. D. Adler Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, G. Hetzer, T. Hrenar, G. Knizia, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, P. Palmieri, K. Pflüger, R. Pitzer, M. Reiher, U. Schumann, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and A. Wolf, MOLPRO, version 2008.1 Search PubMed.
- A. Z. Weller, Elektrochem., 1956, 60, 1144 CAS.
- P. K.Sengupta and M. Kasha, Chem. Phys. Lett., 1979, 68, 382 CrossRef.
- D. McMorrow and M. Kasha, J. Phys. Chem., 1984, 88, 2235 CrossRef CAS.
- Y.-M. Cheng, S.-C. Pu, C.-J. Hsu, C.-H. Lai and P.-T. Chou, ChemPhysChem, 2006, 7, 1372 CrossRef CAS.
- J. Huang, A. Peng, H. Fu, Y. Ma, T. Zhai and J. Yao, J. Phys. Chem. A, 2006, 110, 9079 CrossRef CAS.
- G. Burdzinski, A. Maciejewski, G. Buntinx, O. Poizat and C. Lefumeux, Chem. Phys. Lett., 2003, 368, 745–753 CrossRef CAS.
- G. Burdzinski, M. Ziolek, J. Karolczak and A. Maciejewski, J. Phys. Chem. A, 2004, 108, 11160–11164 CrossRef CAS.
- A. Maciejewski and R. P. Steer, Chem. Rev., 1993, 93, 67–98 CrossRef CAS.
- M. Szymanski, A. Maciejewski and R. P. Steer, Chem. Phys., 1988, 124, 143–154 CrossRef CAS.
- M. Albrecht, C. Bohne, A. Granzhan, H. Ihmels, T. C. S. Pace, A. Schnurpfeil, M. Waidelich and C. Yihwa, J. Phys. Chem. A, 2007, 111, 1036–1044 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |