Graphene oxide-iron oxide and reduced graphene oxide-iron oxide hybrid materials for the removal of organic and inorganic pollutants†
Xin
Yang
,
Changlun
Chen
*,
Jiaxing
Li
,
Guixia
Zhao
,
Xuemei
Ren
and
Xiangke
Wang
*
Key Laboratory of Novel Thin Film Solar Cells, Institute of Plasma Physics, Chinese Academy of Sciences, P. O. Box 1126, Hefei, 230031, P. R. China. E-mail: clchen@ipp.ac.cn; xkwang@ipp.ac.cn.; Fax: +86 5515591310; Tel: +86 551 5592788
First published on 26th July 2012
Abstract
Graphene oxide (GO) and reduced graphene oxide (RGO) were both decorated with iron oxide nanoparticles and were characterized by scanning and transmission electron microscopy, powder X-ray diffraction, Fourier transform infrared spectroscopy, and X-ray photoelectron spectroscopy. The adsorption of Pb(II), 1-naphthol, and 1-naphthylamine, as representatives of inorganic and organic pollutants, on GO-iron oxides and RGO-iron oxides was investigated. The results showed that the GO-iron oxide material was a good adsorbent for Pb(II) but not for 1-naphthol and 1-naphthylamine due to oxygen-containing groups on the surface, whereas the RGO-iron oxide material was a good adsorbent for 1-naphthol and 1-naphthylamine but not for Pb(II). The adsorption of 1-naphthol and 1-naphthylamine on RGO-iron oxides was an endothermic and spontaneous process. Both materials can be easily separated by magnetic separation.
1. Introduction
Graphene, a fascinating two dimensional carbon-based material possessing atomic thickness, has attracted enormous attention from researchers. Owing to its layered structure, graphene has large theoretical specific surface area (∼2630 m2 g−1).1 This superiority allows graphene to be used as a novel adsorbent for environmental pollutant removal. Graphene and graphene oxide (GO) were reported as effective adsorbents toward cationic and anionic dyes.2 GO is a lamellar flexible material with a large number of functional groups such as epoxy (C–O–C), hydroxyl (OH) and carboxyl (COOH) groups on both basal planes and edges.3 One major advantage of GO is that it is hydrophilic with very high negative charge density arising due to the oxygen-containing functional groups. These functional groups are also responsible for the formation of a stable aqueous colloid that is obtained through sonication. In solution phase, GO can act as a weak acid cation exchange resin because of the ionizable carboxyl groups, which allow surface complexation with metal ions or positively charged organic molecules. These functional groups can interact with positively charged species like metal ions,4 polymers,5 and biomolecules,6etc. GO is a layered material with the ability to swell in the presence of water by intercalating water molecules. Ramesha et al.2 observed that GO was excellent material for the adsorption of cationic dyes, while reduced graphene oxide (RGO) worked very well for anionic dyes. In our earlier reports,7,8 few-layered GO and sulfonated graphene nanosheets were synthesized and used to remove persistent aromatic pollutants. These studies reveal that the graphene nanosheets may be promising materials to separate pollutants from wastewater. However, the adsorbent–adsorbate interactions are not studied in detail, and the application of RGO as an adsorbent is still scarce.Although graphene has a high adsorption ability, it is difficult to separate because of its small size. This disadvantage limits its use in reality. Compared with centrifugation and filtration methods, the magnetic separation method is considered as a rapid and effective technique for separating nanoparticles from aqueous solutions.9 The incorporation of magnetite (such as maghemite, magnetite) with graphene or GO may be a promising method. GO has layered nanostructures with high thermal and chemical stabilities, which allow them to function as supports for preparing nanosized metal and metal oxide particle composites. GO has emerged as a precursor offering the potential of cost-effective, large-scale production of graphene-based materials. Graphene-based materials,10–12 and chemically modified graphene including GO have shown many applications in composite materials,13–15 and devices.16–19 Chemical methods offer the potentially low cost and large scale production of graphene-based hybrid materials.20–22 Recently, magnetite-GO and magnetite-graphene hybrids have been synthesized and applied to targeted drug carriers,23 magnetic resonance imaging,24 and pollutant removal.25–28 The large surface area and stability of RGO motivate us to synthesize magnetite-RGO hybrids for pollutant removal. It is interesting to note that GO-iron oxide and RGO-iron oxide hybrid materials have different adsorption capabilities for organic and inorganic pollutants.
Ionizable aromatic compounds such as hydroxyl- and amino-substituted aromatics are found in effluents from the pesticides, dyestuffs, pharmaceuticals, petrochemical, and other industries. Pb(II) is a common type of pollutant in accidental pollutions which are detrimental to humans and living things. The naphthol, naphthylamine and Pb(II) were chosen as representatives of organic and inorganic pollutants.
The objectives of this study were (1) to decorate GO and RGO with iron oxide nanoparticles and to characterize them by scanning electron microscopy (SEM), transmission electron microscopy (TEM), powder X-ray diffraction (XRD), Fourier Transform infrared spectroscopy (FTIR), and X-ray photoelectron spectroscopy (XPS); (2) to describe the adsorption of Pb(II), 1-naphthol, and 1-naphthylamine on GO- iron oxides and RGO-iron oxides; and (3) to elucidate the interaction between graphene and metal ions (or organic molecules) by means of studying the adsorption performance of Pb(II), 1-naphthol and 1-naphthylamine on GO-iron oxides and RGO-iron oxides.
2. Experimental
2.1. Synthesis of GO-iron oxides and RGO-iron oxides
GO was synthesized using a modified Hummers method from natural flake graphite (48 μm, 99.95% purity).29 Briefly, 1.0 g of graphite, 1.0 g of NaNO3, and 40 mL of H2SO4 were mixed and stirred in a three neck flask in an acid bath, and then 6.0 g of KMnO4 was slowly added. Once added, the solution was transferred to a 35 ± 1 °C water bath and stirred for about 1 h, and then 80 mL Milli-Q water was added and the solution was stirred for 30 min at temperature of 90 ± 1 °C. Then, 150 mL Milli-Q water was added, and 6 mL of H2O2 (30%) was added slowly, turning the color of the solution from dark brown to yellow. The warm solution was filtered and rinsed with 100 mL of Milli-Q water. The filter cake was dried under vacuum and a dark brown GO powder was obtained. The synthesis of RGO was from GO reduction with hydrazine hydrate. In brief, GO (0.2 g) was placed in a flask, and then 100 mL water was added, yielding a yellow-brown dispersion. The dispersion was sonicated until it became clear with no visible particulate matter. Hydrazine hydrate (2 mL) was added and the mixture was stirred and heated in an oil bath at 100 °C for 24 h, and then the reduced GO gradually precipitated out as a black solid. The black solid was filtered and rinsed with water and methanol, and then was dried under vacuum.0.15 g GO (or RGO) was first ultrasonicated in 200 mL Milli-Q water to form a homogeneous suspension.30 Then the solution was transferred to a three-neck flask and purged with N2. An aqueous solution of FeCl3·6H2O (0.33 g) and FeSO4·7H2O (0.38 g) in Milli-Q water (5 mL) was injected to GO suspension. The mixture was heated to 80 °C and stirred constantly under N2. Following this, 10 mL 30% ammonia solution was added to adjust the pH to 10 and the mixture was stirred and kept at 80 °C for 30 min. At last, 1.0 g trisodium citrate was added to the solution while the temperature was raised to 95 °C, resulting in a black color suspension. The products were separated with a permanent magnet, rinsed with Milli-Q water several times, and finally dried at 60 °C. GO-iron oxide and RGO-iron oxide hybrid materials were thus obtained.
2.2 Characterization
The morphologies of prepared GO-iron oxide and RGO-iron oxide hybrid materials were characterized by SEM (JEOL JSM-6700, Tokyo, Japan) and TEM (JEOL-2010, Tokyo, Japan). XRD patterns were measured on a X'Pert PRO diffractometer with Cu-Kα radiation (λ = 0.154 nm). FTIR spectra were obtained using a Nicolet 8700 FTIR spectrometer (Thermo Scientific Instrument Co. U.S.A.) equipped with a KBr beam splitter (KBr, FTIR grade) at room temperature. XPS measurements were performed in a VG Scientific ESCALAB Mark II spectrometer equipped with two ultrahigh vacuum chambers. The surface areas were determined by nitrogen adsorption-desorption isotherms (Tristar II 3020 M, Micromeritics Co., USA). The acid–base surface chemistry of GO-iron oxides and RGO-iron oxides was investigated by potentiometric titration experiments.2.3 Adsorption experiments
All adsorption experiments were carried out using a batch equilibrium technique in a serial of 20 mL glass vials equipped with aluminum foil-lined teflon screw caps. Stock suspension of GO-iron oxides or RGO-iron oxides, NaClO4 and Pb(II) (or 1-naphthol, or 1-naphthylamine) were added in the glass vials to achieve the desired concentrations of different components. The pH was adjusted to the needed value by adding negligible volumes (0–20 μL) of 0.1 M HClO4 or NaOH. For example, the pH value of the adsorption isotherm is 6.5 ± 0.1, so a negligible volume of NaOH was added to adjust pH to 6.5 ± 0.1. When we investigated the effect of pH value from 3 to 11, a negligible volume of HClO4 was added to adjust the pH to 3, and a negligible volume of NaOH was added to adjust pH to 11. After the suspensions were shaken for 2 days, the solid and liquid phases were separated by a magnetic process using a permanent magnet made of Nd-Fe-B for analysis of the supernatant solution. The concentration of Pb(II) in the supernatant was analyzed by spectrophotometer at a wavelength of 616 nm by using a Pb(II)-chlorophosphonazo(III) complex. The concentrations of 1-naphthol and 1-naphthylamine in the supernatant were determined using a UV-2550 spectrophotometer at wavelengths of 212 and 213 nm, respectively. The amount of adsorbate adsorbed on the adsorbent was calculated from the difference between the initial concentration and the equilibrium one. All the experimental data were the averages of duplicate determinations, and the relative errors were about 5%.3. Results and discussion
3.1 GO-iron oxide and RGO-iron oxide characterization
The morphology of RGO-iron oxides is shown in Fig. 1A and 1B, and the crumpled silk wave-like graphene sheets and the presence of magnetite nanoparticles suggest that RGO-iron oxide hybrid material is formed. TEM image (Fig. 1B) shows that RGO film is transparent and iron oxide nanoparticles are well dispersed on the surface. Meanwhile, the average size of the iron oxide nanoparticles is about 20 nm which is consistent with the SEM image. From SEM images of GO-iron oxides (Fig. S1†), there is no obvious morphology difference observed.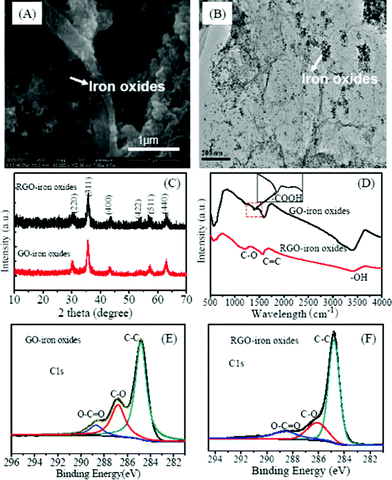 | ||
| Fig. 1 SEM (A), TEM (B), XRD patterns (C), FTIR (D), and high resolution C1s (E, F) XPS of the GO-iron oxide and RGO-iron oxide hybrid materials. | ||
The XRD pattern of RGO-iron oxides is the same as that of GO-iron oxides (Fig. 1C). The main peaks at 2θ = 30.21° (220), 35.71° (311), 43.31° (400), 53.7° (422), 57.35° (511), and 62.72° (440) show the characteristics of iron oxides on both GO-iron oxides and RGO-iron oxides.31 The broad diffraction peaks are indicative of nanoparticles with very small size. Since Fe3O4 has a similar crystal structure to that of γ-Fe2O3, it is hard to distinguish between the two on the basis of the XRD pattern alone. In XRD patterns, there may be two reasons for no peaks from carbon being observed. Firstly, the presence of magnetite reduces the aggregation of graphene sheets, which results in more monolayer graphene, leading to weaker peaks from carbon being observed. Secondly, the strong signals of the iron oxides tend to overwhelm the weak carbon peaks.
The stretching vibrations of epoxy C–O (1225 cm−1), aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (1578 cm−1) and O–H band (3389 cm−1) of H2O are observed both in GO-iron oxides and RGO-iron oxides (Fig. 1D). For GO-iron oxides, there is an extra band around 1399 cm−1 which is due to the vibration of an O–CO group. The peak around 584 cm−1 is attributed to Fe–O, and the enhanced intensity for Fe–O is indicative of the iron load in GO-iron oxides and RGO-iron oxides.31 The wide scan XPS spectra of GO-iron oxides and RGO-iron oxides (Fig. S2†) shows photoelectron lines at the binding energies of about 285, 530, and 711 eV, which are attributed to C 1s, O 1s and Fe 2p, respectively. In the spectra of Fe 2p (Fig. S3†), the peaks Fe 2p3/2 and Fe 2p1/2 are located at 711.29 and 724.82 eV.32 In addition, there is a satellite peak at ∼719.0 eV, characteristic of γ-Fe2O3, which is indicative of the formation of mixed phase of Fe2O3 and Fe3O4 in the GO and RGO matrix.33 Deconvolution of C 1s peaks of GO-iron oxides and RGO-iron oxides are shown in Fig. 1E and 1F. The regions of the spectra can be deconvoluted into three components:34,35 (1) the non-oxygenated C (284.8 eV); (2) the carbon in C–O(∼286.8 eV); and (3) the carboxylate carbon (O–CO, ∼288.8 eV). The intensity of the O–CO peak decreases in the C 1s spectrum of RGO-iron oxides as compared to that of GO-iron oxides. The C/O ratio of GO-iron oxides and RGO-iron oxides is 1.21 and 4.69, respectively. This indicates that oxygen-containing functional groups of RGO-iron oxides are partially reduced. The C1s spectra of raw GO and RGO were also shown in Fig. S4†, the C/O ratio is 2.37 and 4.26.
C (1578 cm−1) and O–H band (3389 cm−1) of H2O are observed both in GO-iron oxides and RGO-iron oxides (Fig. 1D). For GO-iron oxides, there is an extra band around 1399 cm−1 which is due to the vibration of an O–CO group. The peak around 584 cm−1 is attributed to Fe–O, and the enhanced intensity for Fe–O is indicative of the iron load in GO-iron oxides and RGO-iron oxides.31 The wide scan XPS spectra of GO-iron oxides and RGO-iron oxides (Fig. S2†) shows photoelectron lines at the binding energies of about 285, 530, and 711 eV, which are attributed to C 1s, O 1s and Fe 2p, respectively. In the spectra of Fe 2p (Fig. S3†), the peaks Fe 2p3/2 and Fe 2p1/2 are located at 711.29 and 724.82 eV.32 In addition, there is a satellite peak at ∼719.0 eV, characteristic of γ-Fe2O3, which is indicative of the formation of mixed phase of Fe2O3 and Fe3O4 in the GO and RGO matrix.33 Deconvolution of C 1s peaks of GO-iron oxides and RGO-iron oxides are shown in Fig. 1E and 1F. The regions of the spectra can be deconvoluted into three components:34,35 (1) the non-oxygenated C (284.8 eV); (2) the carbon in C–O(∼286.8 eV); and (3) the carboxylate carbon (O–CO, ∼288.8 eV). The intensity of the O–CO peak decreases in the C 1s spectrum of RGO-iron oxides as compared to that of GO-iron oxides. The C/O ratio of GO-iron oxides and RGO-iron oxides is 1.21 and 4.69, respectively. This indicates that oxygen-containing functional groups of RGO-iron oxides are partially reduced. The C1s spectra of raw GO and RGO were also shown in Fig. S4†, the C/O ratio is 2.37 and 4.26.
The nitrogen adsorption-desorption isotherms are given in Fig. S5†. The BET surface area of RGO-iron oxides and GO-iron oxides are 272.59 m2 g−1 and 142.36 m2 g−1, respectively.
The saturation magnetization (Ms) of the GO-iron oxide hybrid material is 31 emu g−1 (magnetic field ± 20 kOe) (Fig. 2), indicating the high magnetic property. The above inset in Fig. 2 is the magnification of hysteresis loop of GO-Fe3O. The Ms of the RGO-iron oxides is the same as that of the GO-iron oxides. The inset of Fig. 2 shows that the GO-iron oxide and RGO-iron oxide hybrid materials are attracted by a magnet, and the clear solution can be easily removed by pipette, or decanted off. This simple magnetic separation experimental result confirms that the GO-iron oxide and RGO-iron oxide hybrid materials are magnetic and can be used as magnetic adsorbents to enrich pollutants from large volumes of aqueous solutions.
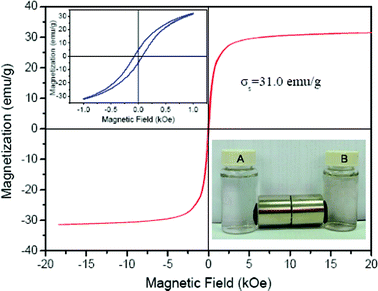 | ||
| Fig. 2 Magnetization curve at room temperature of the GO-iron oxides (inset being separation of particles of GO-iron oxides (A) and RGO-iron oxides (B) by a magnet), indicating that GO-iron oxides has high magnetism. | ||
The surface of adsorbent contains a large number of binding sites. Assuming that surface sites (SOH) are amphoteric, they may become positively charged at low pH due to the protonation reaction on the surfaces (i.e., 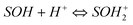 ). At high pH values, the surface of adsorbent surfaces becomes negatively charged due to the deprotonation process (i.e.,
). At high pH values, the surface of adsorbent surfaces becomes negatively charged due to the deprotonation process (i.e., 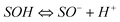 ). Surface site density can be estimated from acid–base titration (described in ESI†). The acid–base titration data for GO-iron oxides and RGO-iron oxides are shown in Fig. 3. TOTH is the total concentration of consumed protons in the titration process, which is calculated from the following equation:
). Surface site density can be estimated from acid–base titration (described in ESI†). The acid–base titration data for GO-iron oxides and RGO-iron oxides are shown in Fig. 3. TOTH is the total concentration of consumed protons in the titration process, which is calculated from the following equation:
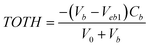 | (1) |
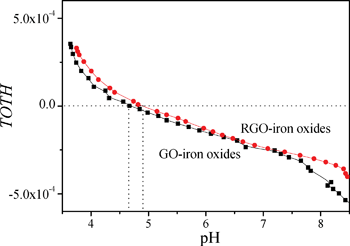 | ||
| Fig. 3 Acid–base titrations of GO-iron oxides and RGO-iron oxides. | ||
3.2 Adsorption isotherms
The adsorption isotherm is most important to understand the distribution of adsorbate molecules between the liquid phase and solid phase when the adsorption process reaches equilibrium. The difference between GO-iron oxides and RGO-iron oxides is the number of surface oxygen-containing functional groups. For the sake of understanding how surface oxygen-containing functional groups affect the adsorption process, we investigated the adsorption isotherms of Pb(II), 1-naphthylamine, and 1-naphthol on GO-iron oxides and RGO-iron oxides (shown in Fig. 4). The experimental data were simulated with the Langmuir (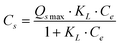 ) and Freundlich (Cs = KF·Ce1/n) models, respectively (where Ce is the equilibrium concentration of adsorbate in aqueous solution (mg L−1), Cs is the amount of adsorbate adsorbed on adsorbent (mg g−1), Qsmax is the maximum amount of adsorbate adsorbed per unit weight of adsorbent to form a complete monolayer coverage on the surface, KL represents enthalpy of sorption and should vary with temperature, KF and n are the Freundlich constants related to the adsorption capacity and sorption intensity, respectively). The relative parameters calculated from the two models are listed in Table 1. The Langmuir model fits the adsorption isotherms better than the Freundlich model, suggesting that the adsorption of Pb(II), 1-naphthylamine, and 1-naphthol on GO-iron oxides and RGO-iron oxides is monolayer coverage. Adsorption capacity of Pb(II) on RGO-iron oxides is much lower than that of Pb(II) on GO-iron oxides, whereas 1-naphthylamine and 1-naphol show the opposite results. The higher adsorption of Pb(II) on GO-iron oxides is attributed to the complexation interaction of Pb(II) with the oxygen-containing groups on GO-iron oxides. The point of zero net proton charge (pHPZNPC) of GO-iron oxides (pHPZNPC = 4.3) is lower than that of RGO-iron oxides (pHPZNPC = 4.8) (Fig. 3) and that the surface of GO-iron oxides has more negative charges than the surface of RGO-iron oxides. The negatively charged surface of GO-iron oxides is in favor of the adsorption of cations due to electrostatic attraction. The qmax values of Pb(II) adsorption on GO-iron oxides and RGO-iron oxides are 588.24 and 454.55 mg g−1, and the qmax values of naphthylamine adsorption on GO-iron oxides and GO-iron oxides are 285.7 and 303.03 mg g−1, respectively. Comparing to qmax values of Pb(II) and naphthylamine adsorption on other adsorbents: activated carbon cloths (42.50 mg g−1 Pb(II) at pH 5 and T = 293 K),36 zinc oxide loading to granular activated carbon (39.40 mg g−1 Pb(II) at pH 5 and T = 298 K),37 carbon nanotubes-iron oxides magnetic composites(54.26 mg g−1 Pb(II) at pH 5 and T = 293 K),38 multiwalled carbon nanotubes (97.08 mg g−1 Pb(II) at pH 5 and T = 298 K),39 multiwalled carbon nanotubes/poly(acrylamide) composite (29.71 mg g−1 Pb(II) at pH 5 and T = 293 K),40 the graphene layer (25.54 mg g−1 Pb(II) at pH 5 and T = 298 K),41 iron oxides (78.1 mg g−1 naphthylamine at pH 6.5 and T = 293 K),42 oxide mutiwalled carbon nanotubes (217.4 mg g−1 naphthylamine at pH 6.5 and T = 293 K),42 multiwall carbon nanotubes/iron oxides, composites (153.8 mg g−1 naphthylamine at pH 6.5 and T = 293 K) 42 multiwall carbon nanotubes/iron oxides/cyclodextrin composite(200.0 mg g−1 naphthylamine at pH 6.5 and T = 293 K).42 It can be seen that the GO-iron oxides and GO-iron oxides hybrid materials have the highest sorption capacity of today's carbon materials.
) and Freundlich (Cs = KF·Ce1/n) models, respectively (where Ce is the equilibrium concentration of adsorbate in aqueous solution (mg L−1), Cs is the amount of adsorbate adsorbed on adsorbent (mg g−1), Qsmax is the maximum amount of adsorbate adsorbed per unit weight of adsorbent to form a complete monolayer coverage on the surface, KL represents enthalpy of sorption and should vary with temperature, KF and n are the Freundlich constants related to the adsorption capacity and sorption intensity, respectively). The relative parameters calculated from the two models are listed in Table 1. The Langmuir model fits the adsorption isotherms better than the Freundlich model, suggesting that the adsorption of Pb(II), 1-naphthylamine, and 1-naphthol on GO-iron oxides and RGO-iron oxides is monolayer coverage. Adsorption capacity of Pb(II) on RGO-iron oxides is much lower than that of Pb(II) on GO-iron oxides, whereas 1-naphthylamine and 1-naphol show the opposite results. The higher adsorption of Pb(II) on GO-iron oxides is attributed to the complexation interaction of Pb(II) with the oxygen-containing groups on GO-iron oxides. The point of zero net proton charge (pHPZNPC) of GO-iron oxides (pHPZNPC = 4.3) is lower than that of RGO-iron oxides (pHPZNPC = 4.8) (Fig. 3) and that the surface of GO-iron oxides has more negative charges than the surface of RGO-iron oxides. The negatively charged surface of GO-iron oxides is in favor of the adsorption of cations due to electrostatic attraction. The qmax values of Pb(II) adsorption on GO-iron oxides and RGO-iron oxides are 588.24 and 454.55 mg g−1, and the qmax values of naphthylamine adsorption on GO-iron oxides and GO-iron oxides are 285.7 and 303.03 mg g−1, respectively. Comparing to qmax values of Pb(II) and naphthylamine adsorption on other adsorbents: activated carbon cloths (42.50 mg g−1 Pb(II) at pH 5 and T = 293 K),36 zinc oxide loading to granular activated carbon (39.40 mg g−1 Pb(II) at pH 5 and T = 298 K),37 carbon nanotubes-iron oxides magnetic composites(54.26 mg g−1 Pb(II) at pH 5 and T = 293 K),38 multiwalled carbon nanotubes (97.08 mg g−1 Pb(II) at pH 5 and T = 298 K),39 multiwalled carbon nanotubes/poly(acrylamide) composite (29.71 mg g−1 Pb(II) at pH 5 and T = 293 K),40 the graphene layer (25.54 mg g−1 Pb(II) at pH 5 and T = 298 K),41 iron oxides (78.1 mg g−1 naphthylamine at pH 6.5 and T = 293 K),42 oxide mutiwalled carbon nanotubes (217.4 mg g−1 naphthylamine at pH 6.5 and T = 293 K),42 multiwall carbon nanotubes/iron oxides, composites (153.8 mg g−1 naphthylamine at pH 6.5 and T = 293 K) 42 multiwall carbon nanotubes/iron oxides/cyclodextrin composite(200.0 mg g−1 naphthylamine at pH 6.5 and T = 293 K).42 It can be seen that the GO-iron oxides and GO-iron oxides hybrid materials have the highest sorption capacity of today's carbon materials.
![Adsorption isotherms of Pb(ii), 1-naphthol and 1-naphthylamine on GO-iron oxides and RGO-iron oxides. m/V = 0.1 g L−1, pH = 6.5 ± 0.1, I = 0.01 M NaClO4. [CPb(ii)]initial = 10∼15 mg L−1, [C1−naphthylamine]initial = 5∼75 mg L−1, [C1−naphthol]initial = 5∼75 mg L−1, T = 303 K. Dots are experimental data and lines are the Langmuir fitting.](/image/article/2012/RA/c2ra20885g/c2ra20885g-f4.gif) | ||
| Fig. 4 Adsorption isotherms of Pb(II), 1-naphthol and 1-naphthylamine on GO-iron oxides and RGO-iron oxides. m/V = 0.1 g L−1, pH = 6.5 ± 0.1, I = 0.01 M NaClO4. [CPb(II)]initial = 10∼15 mg L−1, [C1−naphthylamine]initial = 5∼75 mg L−1, [C1−naphthol]initial = 5∼75 mg L−1, T = 303 K. Dots are experimental data and lines are the Langmuir fitting. | ||
| Experimental conditions | Langmuir | Freundlich | ||||
|---|---|---|---|---|---|---|
| q max (mg g−1) | B (L mg−1) | R 2 | k F (mg1−n Ln g−1) | n | R 2 | |
| Pb(II), GO-iron oxides, 303 K | 588.24 | 0.193 | 0.972 | 168.63 | 0.317 | 0.963 |
| Pb(II), RGO-iron oxides, 303 K | 454.55 | 0.070 | 0.974 | 53.43 | 0.523 | 0.889 |
| 1-naphthol, GO-iron oxides, 303 K | 228.41 | 0.131 | 0.970 | 20.51 | 0.735 | 0.778 |
| 1-naphthol, RGO-iron oxides, 303 K | 243.16 | 0.494 | 0.997 | 162.14 | 0.408 | 0.826 |
| 1-naphthol, RGO-iron oxides, 323 K | 357.00 | 0.252 | 0.994 | 82.66 | 0.419 | 0.700 |
| 1-naphthol, RGO-iron oxides, 343 K | 588.23 | 0.288 | 0.968 | 76.68 | 0.359 | 0.590 |
| 1-naphthylamine, GO-iron oxides, 303 K | 285.71 | 0.177 | 0.985 | 32.34 | 0.534 | 0.894 |
| 1-naphthylamine, RGO-iron oxides, 303 K | 303.03 | 0.589 | 0.997 | 173.73 | 0.134 | 0.893 |
| 1-naphthylamine, RGO-iron oxides, 323 K | 400.00 | 0.481 | 0.986 | 167.68 | 0.236 | 0.972 |
| 1-naphthylamine, RGO-iron oxides, 343 K | 625.00 | 0.593 | 0.990 | 254.74 | 0.315 | 0.787 |
To further understand the Pb(II) adsorption mechanism, the effects of pH and ionic strength on Pb(II) adsorption onto GO-iron oxides were also investigated (Fig. S6†). It turned out that Pb(II) adsorption on GO-iron oxides was strongly dependent on pH values and independent of ionic strength. Meanwhile, the pH values before and after adsorption were measured, and the pH values of the solution after adsorption changed a little to the acidic region (Fig. S7†). Therefore, the high adsorption of Pb(II) on GO-iron oxides was mainly attributed to surface complexation of Pb(II) with the oxygen-containing groups on GO-iron oxides.
Considering the structures of the sp2-bonded carbon atom of graphene, it is expected that the adsorption of organic aromatic compounds on RGO-iron oxides or GO-iron oxides may occur mainly due to a π–π interaction. For GO-iron oxides, there are many more oxygen-containing functional groups on the surface which can act as electron withdrawing groups localizing electron from π system of graphene that might be expected to interfere with π–π dispersion forces between the aromatic ring and graphene. The earlier studies43,44 have shown that the oxygen-containing functional groups depressed the adsorption of organic chemicals on carbon materials by water adsorption, dispersive-repulsive interactions, and hydrogen bonding, thus led to the lower adsorption capacity of GO-iron oxides. Schematics of possible interactions of GO-iron oxides-Pb(II), RGO-iron oxides-naphthol, and RGO-iron oxides-naphthylamine were shown in Fig. S8†.
3.3 Adsorption thermodynamics
The adsorption isotherms of 1-naphthol and 1-naphthylamine on RGO-iron oxides at three different temperatures are shown in Fig. 5. Adsorption capacities are the highest at T = 343 K and are the lowest at T = 303 K, indicating that the adsorption of organic molecules on RGO-iron oxides is promoted at higher temperatures.![Adsorption isotherms and the Langmuir models of 1-naphthylamine and 1-naphthol on RGO-iron oxides at three different temperatures. m/V = 0.1 g L−1, pH = 6.5 ± 0.1, I = 0.01 M NaClO4. [C1−naphthol]initial = 5∼75 mg L−1, [C1−naphthylamine]initial = 5∼75 mg L−1.](/image/article/2012/RA/c2ra20885g/c2ra20885g-f5.gif) | ||
| Fig. 5 Adsorption isotherms and the Langmuir models of 1-naphthylamine and 1-naphthol on RGO-iron oxides at three different temperatures. m/V = 0.1 g L−1, pH = 6.5 ± 0.1, I = 0.01 M NaClO4. [C1−naphthol]initial = 5∼75 mg L−1, [C1−naphthylamine]initial = 5∼75 mg L−1. | ||
The thermodynamic parameters (ΔH0, ΔS0, and ΔG0) for 1-naphthol and 1-naphthylamine adsorption on RGO-iron oxides can be calculated from temperature dependent adsorption isotherms. The values of enthalpy (ΔH0) and entropy (ΔS0) were calculated from the slopes and intercepts of the plot of ln Kdvs. 1/T (Fig. S9) by using the equation ln Kd = ΔS0/R − ΔH0/RT. The Gibbs free energy (ΔG0) of specific adsorption was calculated from the equation ΔG0 = ΔH0 − TΔS0, where R (8.314 J mol−1 K−1) was the ideal gas constant and T/K was the temperature in Kelvin. Relevant calculated data are tabulated in Table 2. The positive values of ΔH0 for 1-naphthol and 1-naphthylamine adsorption indicate that the adsorption of 1-naphthol and 1-naphthylamine on RGO-iron oxides is an endothermic process. The interpretation of the endothermicity of ΔH0 is that 1-naphthol and 1-naphthylamine molecules are solvated in aqueous solution. In order for 1-naphthol and 1-naphthylamine molecules to adsorb on RGO-iron oxides, they have to be denuded their hydration sheath to some extent, and this dehydration process needs energy. The energy of dehydration exceeds the exothermicity of 1-naphthol and 1-naphthylamine molecules to attach to RGO-iron oxides. The negative ΔG0 indicates that the adsorption is a spontaneous process, and the ΔG0 values become more negative with the increase of temperature, indicating that the adsorption process is more favorable at higher temperature due to dehydration of 1-naphthol and 1-naphthylamine. The positive standard entropy change (ΔS0) indicates the fact that the degree of freedom increases at the solid-liquid interface during the adsorption of 1-naphthol and 1-naphthylamine on RGO-iron oxides, and also implies some structural changes in the RGO-iron oxides during adsorption process.
| Experimental conditions | ΔH0 | ΔS0 | ΔG0 |
|---|---|---|---|
| (kJ mol−1) | (J mol−1 K−1) | (kJ mol−1) | |
| 1-naphthol,T = 303 K | 26.27 | 114.02 | −8.343 |
| 1-naphthol, T = 323 K | −10.48 | ||
| 1-naphthol, T = 343 K | −12.92 | ||
| 1-naphtoylamine, T = 303 K | 40.31 | 162.13 | −9.13 |
| 1-naphtoylamine, T = 323 K | −11.41 | ||
| 1-naphtoylamine, T = 343 K | −15.70 |
4. Conclusion
The GO-iron oxides and RGO-iron oxides hybrid materials were synthesized by using GO (or RGO) as matrix. The GO-iron oxides and RGO-iron oxides hybrid materials can be easily separated by magnetic separation. The GO-iron oxides material was a good adsorbent for Pb(II) but not for 1-naphthol and 1-naphthylamine due to oxygen-containing groups on the surface, whereas the RGO-iron oxides material was a good adsorbent for 1-naphthol and 1-naphthylamine but not for Pb(II). The adsorption of 1-naphthol and 1-naphthylamine on RGO-iron oxides was an endothermic and spontaneous process. This work highlighted the interaction of the organic and inorganic pollutants with GO-iron oxides and RGO-iron oxides composites. The results of this research contributed to determining the adsorption performance of inorganic and organic pollutants on GO-iron oxides and RGO-iron oxides composites. Graphene composite materials have enormous potential to rival or even surpass the performance of their carbon nanotube composite counterparts, given that cheap, large-scale production and processing methods for high-quality graphene have become available.Acknowledgements
Financial supports from NSFC (91126020, 21071147, 21071107, 20971126), 973 projects (2011CB933700), and Natural Science Foundation of Anhui Province (Grant No:1208085QB32) are acknowledged.References
- Y. W. Zhu, S. Murali, W. W. Cai, X. S. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS.
- G. K. Ramesha, A. V. Kumara, H. B. Muralidhara and S. Sampath, J. Colloid Interface Sci., 2011, 361, 270–277 CrossRef CAS.
- K. A. Mkhoyan, A. W. Contryman, J. Silcox, D. A. Stewart, G. Eda and C. Mattevi, Nano Lett., 2009, 9, 1058–1063 CrossRef CAS.
- A. K. Mishra and S. Ramaprabhu, J. Hazard. Mater., 2011, 185, 322–328 CrossRef CAS.
- S. T. Yang, Y. L. Chang, H. F. Wang, G. B. Liu, S. Chen and Y. W. Wang, J. Colloid Interface Sci., 2010, 351, 122–127 CrossRef CAS.
- X. L. Zuo, S. L. He, D. Li, C. Peng, Q. Huang, S. P. Song and C. H. Fan, Langmuir, 2010, 26, 1936–1939 CrossRef CAS.
- G. X. Zhao, L. X. Jiang, Y. D. He, J. X. Li, H. L. Dong, X. K. Wang and W. P. Hu, Adv. Mater., 2011, 23, 3959–3963 CrossRef CAS.
- G. X. Zhao, J. X. Li, X. M. Ren, C. L. Chen and X. K. Wang, Environ. Sci. Technol., 2011, 45, 10454–10462 CrossRef CAS.
- C. L. Chen, X. K. Wang and M. Nagatsu, Environ. Sci. Technol., 2009, 43, 2362–2367 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS.
- X. S. Li, W. W. Cai, J. H. An, S. Kim, J. Nah, D. X. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and R. S. Ruff, Science, 2009, 324, 1312–1314 CrossRef CAS.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS.
- T. Ramanathan, A. A. Abdala, S. Stankovich, D. A. Dikin, M. Herrera-Alonso, R. D. Piner, D. H. Adamson, H. C. Schniepp, X. Chen, R. S. Ruoff, S. T. Nguyen, I. A. Aksay, R. K. Prud'Homme and L. L. Brinson, Nat. Nanotechnol., 2008, 3, 327–331 CrossRef CAS.
- G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Malhaüpt, J. Am. Chem. Soc., 2009, 131, 8262–8270 CrossRef CAS.
- F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Nat. Mater., 2007, 6, 652–655 CrossRef CAS.
- W. Y. Kim and K. S. Kim, Nat. Nanotechnol., 2008, 3, 408–412 CrossRef CAS.
- X. Wang, L. I. Zhi and K. Mullen, Nano Lett., 2008, 8, 323–327 CrossRef CAS.
- N. Jung, N. Kim, S. Jockusch, N. J. Turro, P. Kim and L. Brus, Nano Lett., 2009, 9, 4133–4137 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS.
- S. Myung, J. Park, H. Lee, K. S. Kim and S. Hong, Adv. Mater., 2010, 22, 2045–2049 CrossRef CAS.
- X. Y. Yang, X. Y. Zhang, Y. F. Ma, Y. Huang, Y. S. Wang and Y. S. Chen, J. Mater. Chem., 2009, 19, 2710–2714 RSC.
- H. P. Cong, J. J. He, Y. Lu and S. H. Yu, Small, 2009, 6, 169–173 CrossRef.
- M. C. Liu, C. L. Chen, J. Hu, X. L.Wu and X. K. Wang, J. Phys. Chem. C, 2011, 115, 25234–25240 CAS.
- H. M. Sun, L.Y. Cao and L.H. Lu, Nano Res., 2011, 4, 550–562 CrossRef CAS.
- Z. G. Geng, Y. Lin, X. X. Yu, Q.H. Shen, M. Lu, Z. Y. Li, N. Pan and X. P. Wang, J. Mater. Chem., 2012, 22, 3527–3535 RSC.
- G.Q. Xie, P. X. Xi, H. Y. Liu, F. J. Chen, L. Huang, Y. J. Shi, F. P. Hou, Z. Z. Zeng, C. W. Shao and J. Wang, J. Mater. Chem., 2012, 22, 1033–1039 RSC.
- M. Hirata, T. Gotou, S. Horiuchi, M. Fujiwara and M. Ohba, Carbon, 2004, 42, 2929–2937 CAS.
- X. L. Wu, L. Wang, C. L. Chen, A. W. Xu and X. K. Wang, J. Mater. Chem., 2011, 21, 17353–17359 RSC.
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I. C. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979–3986 CrossRef CAS.
- J. Lu, X. L. Jiao, D. R. Chen and W. Li, J. Phys. Chem. C, 2009, 113, 4012–4017 CAS.
- D. H. Zhang, Z. Q. Liu, S. Han, C. Li, B. Lei and M. P. Stewart, Nano Lett., 2004, 4, 2151–2155 CrossRef CAS.
- K. Zhang, L. Mao, L. L. Zhang, H. S. O. Chan, X. S. Zhao and J. S. Wu, J. Mater. Chem., 2011, 21, 7302–7307 RSC.
- Y. Wang, Y. Y. Shao, D. W. Matson, J. H. Li and Y. H. Lin, ACS Nano, 2010, 4, 1790–1798 CrossRef CAS.
- K. Kadirvelu, C. Fau-Brasquet and P. Le Cloirec, Langmuir, 2000, 16, 8404–8409 CrossRef CAS.
- Y. Kikuchi, Q. R. Qian, M. Machida and H. Tatsumoto, Carbon, 2006, 44, 195–202 CrossRef CAS.
- X. J. Peng, Z. K. Luan, Z. C. Di, Z. G. Zhang and C. L. Zhu, Carbon, 2005, 43, 855–894 CrossRef.
- Y. H. Li, J. Ding, Z. K. Luan, Z. C. Di, Y. F. Zhu, C. L. Xu, D. H. Wu and B. Q. Wei, Carbon, 2003, 41, 2787–2792 CrossRef CAS.
- S. B. Yang, J. Hu, C. L. Chen, D. D. Shao and X. K. Wang, Environ. Sci. Technol., 2011, 45, 3621–3627 CrossRef CAS.
- M. Machida, T. Mochimaru and H. Tatsumoto, Carbon, 2006, 44, 2681–2688 CrossRef CAS.
- J. Hu, D. D. Shao, C. L. Chen, G. D. Sheng, X. M. Ren and X. K. Wang, J. Hazard. Mater., 2011, 185, 463–471 CrossRef CAS.
- M. Franz, H. A. Arafat and N. G. Pinto, Carbon, 2000, 38, 1807–1819 CrossRef CAS.
- D. Q. Zhu and J. J. Pignatello, Environ. Sci. Technol., 2005, 39, 2033–2041 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: More characterization of the GO-iron oxide and RGO-iron oxide composites, effect of pH and ionic strength on Pb(II) adsorption onto GO-iron oxides, possible interaction mechanisms of GO-iron oxides-Pb(II), RGO-iron oxides-naphthol, and RGO-iron oxides-naphthylamine, linear plots of lnK0vs. 1/T. See DOI: 10.1039/c2ra20885g/ |
| This journal is © The Royal Society of Chemistry 2012 |