A route to hydroxylfluorenes: TsOH-mediated condensation reactions of 1,3-diketones with propargylic alcohols†
Liang-Feng
Yao
,
Davin
Tan
,
Xiaohe
Miao
and
Kuo-Wei
Huang
*
King Abdullah University of Science and Technology (KAUST), Division of Chemical and Life Sciences and Engineering and KAUST Catalysis Center, Thuwal 23955-6900, Saudi Arabia. E-mail: hkw@kaust.edu.sa
First published on 24th July 2012
Abstract
An efficient method of preparing hydroxylfluorenes by TsOH-mediated tandem alkylation/rearrangements of propargylic alcohols with 1,3-diketones is described. These reactions are accomplished in moderate to good yields under mild conditions to offer a straightforward and convenient one step synthetic route to hydroxylfluorene derivatives through a plausible mechanism involving a sequence of dehydration, addition, rearrangement and aromatization.
Introduction
Fluorenes represent an important class of core structure in organic chemistry because of their wide applications. For example, fluorenes are present in advanced materials possessing unique electronic and photonic properties1–5 that can be used as organic light-emitting diodes (OLEDs),6–12 thin film transistors,13–15 photovoltaic cells,16–20etc., and as versatile building blocks for functional materials,21–26 such as microporous systems for gas storage. The most common methods for the construction of fluorenes include Friedel–Crafts ring closure of biaryl-2-yl-methanols in the presence of acids at high temperature,27 addition of organometallic reagents to 9-fluorenones,28 Friedel–Crafts alkylation of fluorenes,29,30 Pd-catalyzed annulative reaction of dihalobenzenes with hindered Grignard reagents,31 metal-catalyzed or mediated reactions involving Pd-catalyzed rearrangement reactions,32 activation of C–F/C–H bonds of o-arylated α,α,α-trifluorotoluene derivatives,33etc. Although these methods are effective, the development of new economical and convenient synthetic routes for the synthesis of fluorene derivatives remains a major goal for modern organic synthesis to enable a broader substrate scope of the starting materials.Up to date, there have been some studies in which unsaturated alcohols were well explored.34–39 In addition, recent reports have also demonstrated the use of propargylic alcohols as viable substrates in several carbon–carbon and carbon–heteroatom bond formation tactics, normally catalyzed by Lewis and Brønsted acids.40–55 Despite the novelty and success of these methods, preparation of poly-substituent fluorenes from readily available propargyl alcohols remains unknown. Mild and efficient methodologies for the formation of this class of carbocycles from readily accessible substrates in a one pot reaction would be desirable. Herein, we report the preparation of 3-hydroxyfluorene derivatives from a Brønsted acid-catalyzed reaction of propargylic alcohols with 1,3-diketones under mild conditions.
Results and discussion
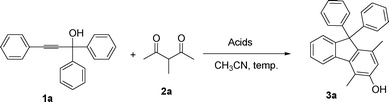
Initial investigations were performed using 1,1,3-triphenylprop-2-yn-1-ol 1a and 3-methylpentane-2,4-dione 2a as substrates in the presence of Brønsted acids (TsOH, MsOH) in CH3CN (acetonitrile). In contrast to the literature result where 1a reacts with 2a in the presence of a Brønsted acid through a regioselective allenylation reaction to afford an allene derivative, we have now demonstrated that compound 3a could be achieved by slightly elevating the reaction temperature to 60 °C. Its molecular structure containing a fluorene skeleton was confirmed by single crystal X-ray crystallography (Fig. 1).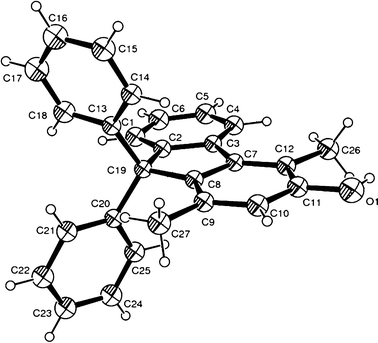 | ||
| Fig. 1 The molecular structure of 3a with thermal ellipsoids at 30% probability levels. | ||
These encouraging observations prompted us to further optimize the reaction conditions and to extend the reaction scope. The experimental results are summarized in Table 1. The influence of the amount of acid and the reaction temperature on the reaction yields were examined. When the reaction was carried out with 0.1 equivalents of TsOH at 60 °C, the corresponding product 3a was obtained in 60% yield (entry 1). By increasing the amount of TsOH, the yield of 3a could be improved to 64–75% (entries 2, 3 and 4). However, when the reaction was carried out at room temperature, only a trace amount of 3a was obtained (entry 5). The yield of the product was slightly reduced when MsOH was used as the catalyst instead (entry 6). By changing the ratio of the starting materials 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a from 1:1.2 to 1.2:1, a slightly lower yield was obtained (entry 7).
:2a from 1:1.2 to 1.2:1, a slightly lower yield was obtained (entry 7).
|
|
|||
|---|---|---|---|
| Entrya | Acids | Temp. | Yield of 3ab |
|
a All of reactions were carried out with 1a (0.3 mmol), 2a (0.36 mmol) in CH3CN (3 mL).
b Isolated yields.
c The ratio of 1a:2a was 1.2:1.
|
|||
| 1 | TsOH (0.1 eq) | 60 °C | 60% |
| 2 | TsOH (0.5 eq) | 60 °C | 64% |
| 3 | TsOH (1.0 eq) | 60 °C | 75% |
| 4 | TsOH (2.0 eq) | 60 °C | 67% |
| 5 | TsOH (0.1 eq) | rt | trace |
| 6 | MsOH (1.0 eq) | 60 °C | 65% |
| 7 | TsOH (1.0 eq) | 60 °C | 52%c |
Under the optimized reaction conditions (entry 3 of Table 1), the synthesis of 3-hydroxyfluorene derivatives was carried out with a variety of commercially available and synthesized propargylic alcohols (Table 2). Reactions of propargylic alcohols with either electron-withdrawing or electron-donating substituents on the aromatic ring of the carbinol and/or alkyne all proceeded well and afforded the desired 3-hydroxyfluorene products in good to moderate yields. Lower yields, however, were obtained when bulky reactants/substituents were employed (entries 6 and 8), suggesting the reaction is sensitive to steric effects. In the case of substrate 1j, where a naphthyl group was attached to a terminal alkyne, the corresponding product 3j was produced in 63% yield (entry 9). The reaction of alcohol 1k with 2a also proceeded properly and furnished the desired product 3k in 58% yield (entry 10). Gram scale synthesis of 3a was also carried out with 68% yield (Scheme 1).
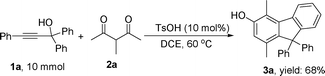 | ||
| Scheme 1 Reaction of 1a with 2a on the gram scale. | ||
|
|
|||
|---|---|---|---|
| Entry | R1/R2 | R3 | Yield of 3a |
| a Isolated yields. | |||
| 1 | 4-Me/H, 1b | Me, 2a | 3b, 70% |
| 2 | 3,5-F2/H, 1c | Me, 2a | 3c, 62% |
| 3 | 4-MeO/H, 1d | Me, 2a | 3d, 66% |
| 4 | 4-CI/H, 1e | Me, 2a | 3e, 67% |
| 5 | 3,5-Me2/H, 1f | Me, 2a | 3f, 50% |
| 6 | 2-Me/H, 1g | Me, 2a | 3g, 26% |
| 7 | H/Me, 1h | Me, 2a | 3h, 66% |
| 8 | H/H, 1a | Bn, 2b | 3i, 30% |
| 9 |
|
Me, 2a | 3j, 63% |
| 10 |
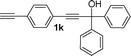
|
Me, 2a | 3k 58% |
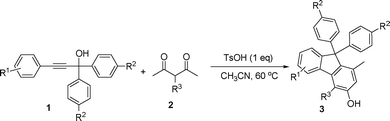
In order to have a better understanding of the reaction mechanism, we carried out a control experiment by employing 4a56 as the substrate under identical reaction conditions. The reaction afforded the corresponding product 3a in 86% yield, indicating that compound 4a could be an intermediate for this tandem multistep reaction (Scheme 2). Additional information was provided by 2D NOESY analysis on compound 3i, where the methyl group was confirmed to be in the meta-position to the hydroxyl group (Fig. 2 and ESI†). This observation suggests that the C–C bond cleavage may occur between C2 and C3 of 3-subsituted pentane-2,4-dione, followed by a cyclization reaction. Accordingly, a plausible reaction mechanism was proposed in Scheme 3. The first step involves the reaction of 1 with 2 to produce intermediate 4via a nucleophilic addition of the 1,3-diketone to the allenic cation.56 Subsequently, 4 can undergo an intramolecular electrophilic substitution of one of the benzene rings via a Wheland-type intermediate A,57,58 which needs thermal input to take place, to produce intermediate B. The highly strained 4-membered ring B may readily undergo the ring opening reaction (retro-aldol) to form intermediate C, a process analogous to the DeMayo reaction.59 Subsequent enol–keto tautomerization reactions form intermediate E, to allow an intramolecular aldol condensation to the cyclic enone G.60 The reaction is then concluded by the aromatization (tautomerization) of G to generate the final product.61
 | ||
| Fig. 2 NOE correlations for 3i. | ||
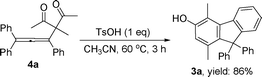 | ||
| Scheme 2 Control experiment of reaction with 4a. | ||
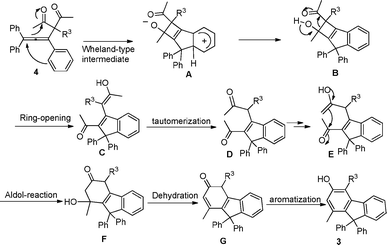 | ||
| Scheme 3 A suggested reaction mechanism for the PTSA-mediated reaction of 1a with 2a. | ||
Moreover, we tried to examine the further transformation of 3avia deoxygenation of the phenol group in 3a. Initially, 3a can be transformed to its corresponding triflate 3a′, which is readily prepared.62 However, when we tested the deoxygenation of 3avia its triflate using two established reports,63,64 no reaction occurred.
Conclusions
In summary, we have demonstrated an efficient and novel TsOH-mediated synthetic route to poly-substituted hydroxylfluorene based on a tandem reaction of propargylic alcohols with 1,3-diketones. This reaction is proposed via a sequence of dehydration, addition, rearrangement and aromatization. The reaction protocol is simple and scalable, and it generates the desired products from readily available and low cost starting materials in good yields, with H2O as the only by-product. Efforts are currently under way to apply the method to natural product synthesis and will be reported in due course.Experimental section
General remarks
1H NMR and 13C NMR spectra were recorded on a 400 or 500 MHz spectrometer in CDCl3. HRMS was measured on a Thermo Analytical Concept mass spectrometer (ESI). Melting points are uncorrected. All reactions were monitored by TLC with silica gel coated plates. Flash column chromatography was carried out using 300–400 mesh silica gel.:ethyl acetate = 10:1) to give the title compound.
3-(3,5-Difluorophenyl)-1,1-diphenylprop-2-yn-1-ol (1c)
A colorless oil; 1H NMR (CDCl3, 500 MHz, TMS) δ 3.50 (s, 1H, OH), 6.88–6.92 (m, 1H, Ar), 7.09–7.10 (m, 2H, Ar), 7.38–7.48 (m, 6H, Ar), 7.72–7.88 (m, 4H, Ar). 13C NMR (CDCl3, 125 MHz, TMS) δ 74.7, 84.7, 93.6, 104.6 (d, JC–F = 25 Hz), 104.8 (d, JC–F = 25 Hz), 114.5 (d, JC–F = 21.6 Hz), 114.6 (d, JC–F = 21.6 Hz), 124.8 (d, JC–F = 11.8 Hz). 124.9 (d, JC–F = 11.6 Hz), 125.9, 127.8, 128.3, 144.3, 162.4 (d, JC–F = 247.5 Hz), 162.5 (d, JC–F = 247.5 Hz). MS (%) ESI 321 [M + H]+. HRMS (ESI) calcd. for C21H15F2O: 321.1091, found: 321.1076.3-(3,5-Dimethylphenyl)-1,1-diphenylprop-2-yn-1-ol (1f)
A colorless oil; 1H NMR (CDCl3, 400 MHz, TMS) δ 2.30 (s, 6H, 2CH3), 2.86 (s, 1H, OH), 6.99 (s, 1H, Ar), 7.16 (s, 2H, Ar), 7.29 (d, 2H, J = 7.2 Hz, Ar), 7.34–7.37 (m, 4H, Ar), 7.67–7.69 (m, 4H, Ar). 13C NMR (CDCl3, 100 MHz, TMS) δ 21.1, 74.8, 87.6, 90.9, 122.0, 126.0, 127.7, 128.3, 129.4, 130.6, 137.9, 145.1. MS (%) ESI 311 [M − H]−. HRMS (ESI) calcd. for C23H19O: 311.1436, found: 311.1446.1,4-Dimethyl-9,9-diphenyl-9H-fluoren-3-ol (3a)
A white solid, m.p. 203–205 °C; 1H NMR (CDCl3, 400 MHz, TMS) δ 1.91 (s, 3H, CH3), 2.71 (s, 3H, CH3), 4.86 (br, 1H, OH), 6.53 (s, 1H, Ar), 7.21–7.41 (m, 13H, Ar), 7.99 (d, 2H, J = 7.8 Hz, Ar). 13C NMR (CDCl3, 100 MHz, TMS) δ 12.3, 19.9, 64.5, 116.8, 117.1, 123.2, 125.4, 126.4, 127.0, 127.3, 127.9, 129.1, 133.8, 140.4, 141.0, 142.4, 142.6, 153.4, 155.7. MS (%) ESI 361 [M − H]−. HRMS (ESI) calcd. for C27H21O: 361.1592, found: 361.1596.1,4,7-Trimethyl-9,9-diphenyl-9H-fluoren-3-ol (3b)
A white solid, m.p. 253–255 °C; 1H NMR (CDCl3, 400 MHz, TMS) δ 1.89 (s, 3H, CH3), 2.33 (s, 3H, CH3), 2.68 (s, 3H, CH3), 4.75 (br, 1H, OH), 6.50 (s, 1H, Ar), 7.15–7.34 (m, 12H, Ar), 7.86 (d, 2H, J = 7.9 Hz, Ar). 13C NMR (CDCl3, 100 MHz, TMS) δ 12.2, 19.9, 21.6, 64.3, 116.4, 116.6, 122.9, 126.0, 126.3, 127.8, 128.0, 129.1, 133.7, 137.2, 138.3, 140.5, 142.3, 142.8, 153.3, 155.9. MS (%) ESI 375 [M − H]−. HRMS (ESI) calcd. for C28H23O: 375.1749, found: 375.1782.6,8-Difluoro-1,4-dimethyl-9,9-diphenyl-9H-fluoren-3-ol (3c)
A white solid, m.p. 222–224 °C ; 1H NMR (CDCl3, 400 MHz, TMS) δ 1.85 (s, 3H, CH3), 2.66 (s , 3H, CH3), 4.78 (br, 1H, OH), 6.57 (s, 1H, Ar), 6.65 (td, 1H, J1 = 9.4 Hz, J2 = 2.1 Hz, Ar), 7.27–7.32 (m, 6H, Ar), 7.37–7.39 (m, 4H, Ar), 7.54 (dd, 1H, J1 = 9.4 Hz, J2 = 2.0 Hz, Ar). 13C NMR (CDCl3, 100 MHz, TMS) δ 12.0, 19.8, 63.5, 102.7 (d, JC–F = 26.0 Hz), 102.9 (d, JC–F = 26.0 Hz), 106.6 (d, JC–F = 23.9 Hz), 106.7 (d, JC–F = 23.9 Hz), 117.5, 118.2, 126.8, 127.9, 128.5, 128.8, 128.9, 133.9, 136.3 (d, JC–F = 3.0 Hz), 136.4 (d, JC–F = 3.0 Hz), 138.7, 139.9, 143.5, 144.5 (d, JC–F = 6.9 Hz), 144.6 (d, JC–F = 6.9 Hz), 153.4, 157.8 (d, JC–F = 249.6 Hz), 158.0 (d, JC–F = 249.6 Hz), 162.9 (d, JC–F = 243.7 Hz), 163.0 (d, JC–F = 243.7 Hz) . MS (%) ESI 397 [M − H]−. HRMS (ESI) calcd. for C27H19F2O: 397.1404, found: 397.1439.7-Methoxy-1,4-dimethyl-9,9-diphenyl-9H-fluoren-3-ol (3d)
A white solid, m.p. 251–253 °C; 1H NMR (CDCl3, 400 MHz, TMS) δ 1.89 (s, 3H, CH3), 2.66 (s, 3H, CH3), 3.78 (s, 3H, OCH3), 4.86 (br, 1H, OH), 6.46 (s, 1H, Ar), 6.88–6.93 (m, 2H, Ar), 7.22–7.34 (m, 10H, Ar), 7.89 (d, 1H, J = 8.4 Hz, Ar). 13C NMR (CDCl3, 100 MHz, TMS) δ 12.1, 19.8, 55.3, 64.4, 111.4, 112.5, 115.7, 116.0, 123.8, 126.3, 127.8, 129.0, 133.5, 133.9, 140.3, 141.9, 142.6, 153.3, 157.5, 159.1. MS (%) ESI 391 [M − H]−. HRMS (ESI) calcd. for C28H23O2: 391.1698, found: 391.1741.7-Chloro-1,4-dimethyl-9,9-diphenyl-9H-fluoren-3-ol (3e)
A white solid, m.p. 204–206 °C; 1H NMR (CDCl3, 400 MHz, TMS) δ 1.94 (s, 3H, CH3), 2.36 (s, 6H, 2CH3), 2.72 (s, 3H, CH3), 4.83 (br, 1H, OH), 6.53 (s, 1H, Ar), 7.09 (d, 4H, J = 8.1 Hz, Ar), 7.22–7.25 (m, 5H, Ar), 7.31–7.37 (m, 2H, Ar), 7.41 (d, 1H, J = 7.6 Hz, Ar). 13C NMR (CDCl3, 100 MHz, TMS) δ 12.2, 19.8, 20.9, 63.8, 116.7, 116.9, 123.0, 125.2, 126.8, 127.2, 128.5, 128.9, 133.6, 135.8, 139.6, 140.3, 140.8, 142.6, 153.1, 155.9. MS (%) ESI 389 [M − H]−. HRMS (ESI) calcd. for C29H25O: 389.1905, found: 389.1942.1,4,6,8-Tetramethyl-9,9-diphenyl-9H-fluoren-3-ol (3f)
A white solid, m.p. 267–269 °C; 1H NMR (CDCl3, 400 MHz, TMS) δ 1.88 (s, 3H, CH3), 2.67 (s, 3H, CH3), 4.80 (br, 1H, OH), 6.54 (s, 1H, Ar), 7.27–7.35 (m, 12H, Ar), 7.89 (d, 1H, J = 8.3 Hz, Ar). 13C NMR (CDCl3, 100 MHz, TMS) δ 12.2, 19.8, 64.4, 117.0, 117.1, 124.0, 125.7, 126.6, 127.3, 128.0, 129.0, 132.9, 133.8, 139.3, 139.5, 141.8, 142.2, 153.4, 157.3. MS (%) ESI 395 [M − H]−. HRMS (ESI) calcd. for C27H20ClO: 395.1203, found: 395.1240.1,4,5-Trimethyl-9,9-diphenyl-9H-fluoren-3-ol (3g)
A white solid, m.p. 260–262 °C ; 1H NMR (CDCl3, 400 MHz, TMS) δ 1.82 (s, 3H, CH3), 1.89 (s, 3H, CH3), 2.41 (s, 3H, CH3), 2.67 (s, 3H, CH3), 4.82 (br, 1H, OH), 6.39 (s, 1H, Ar), 6.79 (s, 1H, Ar), 7.16–7.26 (m, 6H, Ar), 7.36–7.39 (m, 4H, Ar), 7.72 (s, 1H, Ar). 13C NMR (CDCl3, 100 MHz, TMS) δ 12.4, 20.0, 20.2, 21.5, 64.5, 116.5, 117.1, 122.0, 126.2, 127.6, 129.1, 131.1, 132.6, 134.3, 136.9, 140.1, 140.2, 142.0, 145.3, 150.2. MS (%) ESI 389 [M − H]−. HRMS (ESI) calcd. for C29H25O: 389.1905, found: 389.1943.1,4-Dimethyl-9,9-dip-tolyl-9H-fluoren-3-ol (3h)
A yellow oil; 1H NMR (CDCl3, 400 MHz, TMS) δ 1.63 (s, 3H, CH3), 1.90 (s, 3H, CH3), 2.56 (s, 3H, CH3), 2.67 (s, 3H, CH3), 4.83 (br, 1H, OH), 6.57 (s, 1H, Ar), 7.13–7.25 (m, 8H, Ar), 7.27–7.33 (m, 5H, Ar). 13C NMR (CDCl3, 100 MHz, TMS) δ 16.8, 20.0, 24.4, 64.6, 116.2, 116.6, 122.7, 126.3, 127.4, 127.7, 129.1, 130.6, 131.7, 133.6, 139.5, 142.5, 142.6, 142.9, 154.4, 156.2. MS (%) ESI 375 [M − H]−. HRMS (ESI) calcd. for C28H23O: 375.1749, found: 375.1789.4-Benzyl-1-methyl-9,9-dip-tolyl-9H-fluoren-3-ol (3i)
A yellow solid, m.p. 152–154 °C; 1H NMR (CDCl3, 400 MHz, TMS) δ 1.95 (s, 3H, CH3), 4.62 (s, 2H, CH2), 4.78 (br, 1H, OH), 6.60 (s, 1H, Ar), 7.15–7.33 (m, 18H, Ar), 7.34–7.35 (m, 1H, Ar). 13C NMR (CDCl3, 100 MHz, TMS) δ 20.1, 31.6, 64.4, 117.3, 119.1, 122.9, 125.4, 126.2, 126.4, 127.2, 127.5, 127.9, 128.1, 128.7, 129.1, 135.1, 139.1, 140.1, 140.9, 142.5, 142.8, 154.1. MS (%) ESI 437 [M − H]−. HRMS (ESI) calcd. for C33H25O: 437.1905, found: 437.1942.8,11-Dimethyl-7,7-dip-tolyl-7H-benzo[c]fluoren-10-ol (3j)
A white solid, m.p. 265–267 °C; 1H NMR (CDCl3, 500 MHz, TMS) δ 1.92 (s, 3H, CH3), 2.65 (s, 3H, CH3), 4.87 (s, 1H, OH), 6.55 (s, 1H, Ar), 7.16–7.22 (m, 6H, Ar), 7.33–7.34 (m, 4H, Ar), 7.43–7.45 (m, 2H, Ar), 7.50–7.53 (m, 1H, Ar), 7.68 (d, 1H, J = 8.5 Hz, Ar), 7.81 (d, 1H, J = 8.5 Hz, Ar). 8.32 (d, 1H, J = 8.5 Hz, Ar). 13C NMR (CDCl3, 125 MHz, TMS) δ 17.4, 19.9, 65.4, 116.1, 116.4, 122.9, 124.7, 125.0, 126.5, 127.2, 127.9, 128.2, 128.3, 129.0, 133.6, 133.7, 136.0, 141.6, 142.1, 143.0, 153.7, 154.9. MS (%) ESI 411 [M − H]−. HRMS (ESI) calcd. for C31H23O: 411.1749, found: 411.1797.7-Ethynyl-1,4-dimethyl-9,9-dip-tolyl-9H-fluoren-3-ol (3k)
A white solid, m.p. 231–233 °C; 1H NMR (CDCl3, 500 MHz, TMS) δ 1.85 (s, 3H,CH3), 2.65 (s, 3H, CH3), 3.04 (s, 1H, CCH), 4.81 (s, 1H, OH), 6.51 (s, 1H, Ar), 7.21–7.28 (m, 10H, Ar), 7.44 (s, 1H, Ar), 7.45 (d, 1H, J = 8.0 Hz, Ar), 7.90 (d, 1H, J = 8.0 Hz, Ar). 13C NMR (CDCl3, 125 MHz, TMS) δ 12.2, 19.8, 62.5, 84.2, 117.3, 117.4, 120.4, 122.9, 126.5, 127.9, 128.96, 129.01, 131.2, 133.8, 139.5, 141.6, 141.9, 142.7, 153.4, 155.5. MS (%) ESI 385 [M − H]−. HRMS (ESI) calcd. for C29H21O: 385.1592, found: 385.1640.Acknowledgements
We would like to acknowledge the financial support from the King Abdullah University of Science and Technology and the assistance given by the Core Laboratories.References
- A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS.
- A. Facchetti, Chem. Mater., 2010, 23, 733–758 CrossRef.
- U. Giovanella, P. Betti, C. Botta, S. Destri, J. Moreau, M. Pasini, W. Porzio, B. Vercelli and A. Bolognesi, Chem. Mater., 2010, 23, 810–816 CrossRef.
- L. Luo, S. H. Choi and C. D. Frisbie, Chem. Mater., 2010, 23, 631–645 CrossRef.
- H. Yao, H. Sasahara and K. Kimura, Chem. Mater., 2011, 23, 913–922 CrossRef CAS.
- A. L. Fisher, K. E. Linton, K. T. Kamtekar, C. Pearson, M. R. Bryce and M. C. Petty, Chem. Mater., 2011, 23, 1640–1642 CrossRef CAS.
- T. Hadizad, J. Zhang, Z. Y. Wang, T. C. Gorjanc and C. Py, Org. Lett., 2005, 7, 795–797 CrossRef CAS.
- S. Merlet, M. Birau and Z. Y. Wang, Org. Lett., 2002, 4, 2157–2159 CrossRef CAS.
- U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477–487 CrossRef CAS.
- D. Thirion, J. l. Rault-Berthelot, L. Vignau and C. Poriel, Org. Lett., 2011, 13, 4418–4421 CrossRef CAS.
- K.-T. Wong, L.-C. Chi, S.-C. Huang, Y.-L. Liao, Y.-H. Liu and Y. Wang, Org. Lett., 2006, 8, 5029–5032 CrossRef CAS.
- K.-T. Wong, T.-Y. Hwu, A. Balaiah, T.-C. Chao, F.-C. Fang, C.-T. Lee and Y.-C. Peng, Org. Lett., 2006, 8, 1415–1418 CrossRef CAS.
- D. Katsis, Y. H. Geng, J. J. Ou, S. W. Culligan, A. Trajkovska, S. H. Chen and L. J. Rothberg, Chem. Mater., 2002, 14, 1332–1339 CrossRef CAS.
- J.-H. Kim, H. U. Kim, D. Mi, S.-H. Jin, W. S. Shin, S. C. Yoon, I.-N. Kang and D.-H. Hwang, Macromolecules, 2012, 45, 2367–2376 CrossRef CAS.
- W. Wu, Y. Liu and D. Zhu, Chem. Soc. Rev., 2010, 39, 1489–1502 RSC.
- X.-Y. Cao, W.-B. Zhang, J.-L. Wang, X.-H. Zhou, H. Lu and J. Pei, J. Am. Chem. Soc., 2003, 125, 12430–12431 CrossRef CAS.
- F. Jaramillo-Isaza and M. L. Turner, J. Mater. Chem., 2006, 16, 83–89 RSC.
- C. Müller, E. Wang, L. M. Andersson, K. Tvingstedt, Y. Zhou, M. R. Andersson and O. Inganäs, Adv. Funct. Mater., 2010, 20, 2124–2131 CrossRef.
- H. Reisch, U. Wiesler, U. Scherf and N. Tuytuylkov, Macromolecules, 1996, 29, 8204–8210 CrossRef CAS.
- R. Verduzco, I. Botiz, D. L. Pickel, S. M. Kilbey, K. Hong, E. Dimasi and S. B. Darling, Macromolecules, 2011, 44, 530–539 CrossRef CAS.
- C.-S. Li, Y.-H. Tsai, W.-C. Lee and W.-J. Kuo, J. Org. Chem., 2010, 75, 4004–4013 CrossRef CAS.
- Z. H. Li and M. S. Wong, Org. Lett., 2006, 8, 1499–1502 CrossRef CAS.
- K. M. Omer, S.-Y. Ku, K.-T. Wong and A. J. Bard, Angew. Chem., Int. Ed., 2009, 48, 9300–9303 CrossRef CAS.
- J. Weber and A. Thomas, J. Am. Chem. Soc., 2008, 130, 6334–6335 CrossRef CAS.
- J.-S. Yang, H.-H. Huang, Y.-H. Liu and S.-M. Peng, Org. Lett., 2009, 11, 4942–4945 CrossRef CAS.
- F. Moreau, N. Audebrand, C. Poriel, V. Moizan-Basle and J. Ouvry, J. Mater. Chem., 2011, 21, 18715–18722 RSC.
- T. Iihama, J. m. Fu, M. Bourguignon and V. Snieckus, Synthesis, 1989, 1989, 184 CrossRef.
- M. Orfanopoulos and I. Smonou, Synth. Commun., 1988, 18, 833–839 CrossRef CAS.
- M. Bruch, M. Große and D. Rewicki, Justus Liebigs Ann. Chem., 1976, 1976, 74–88 CrossRef.
- J. F. Cairns and W. J. Hickinbottom, J. Chem. Soc., 1962, 867–870 RSC.
- C.-G. Dong and Q.-S. Hu, Angew. Chem., Int. Ed., 2006, 45, 2289–2292 CrossRef CAS.
- Q. Tian and R. C. Larock, Org. Lett., 2000, 2, 3329–3332 CrossRef CAS.
- K. Fuchibe and T. Akiyama, J. Am. Chem. Soc., 2006, 128, 1434–1435 CrossRef CAS.
- M. Bandini and M. Tragni, Org. Biomol. Chem., 2009, 7, 1501–1507 CAS.
- B. Biannic and A. Aponick, Eur. J. Org. Chem., 2011, 2011, 6605–6617 CrossRef CAS.
- E. Emer, R. Sinisi, M. G. Capdevila, D. Petruzziello, F. De Vincentiis and P. G. Cozzi, Eur. J. Org. Chem., 2011, 2011, 647–666 CrossRef.
- N. Ljungdahl and N. Kann, Angew. Chem., Int. Ed., 2009, 48, 642–644 CrossRef CAS.
- J. Muzart, Tetrahedron, 2005, 61, 4179–4212 CrossRef CAS.
- J. Muzart, Tetrahedron, 2008, 64, 5815–5849 CrossRef CAS.
- P. Kothandaraman, S. R. Mothe, S. S. M. Toh and P. W. H. Chan, J. Org. Chem., 2011, 76, 7633–7640 CrossRef CAS.
- P. Kothandaraman, W. Rao, S. J. Foo and P. W. H. Chan, Angew. Chem., Int. Ed., 2010, 49, 4619–4623 CrossRef CAS.
- S. R. Mothe, P. Kothandaraman, S. J. L. Lauw, S. M. W. Chin and P. W. H. Chan, Chem. Eur. J., 2012, 18, 6133–6137 CrossRef CAS.
- S. R. Mothe, P. Kothandaraman, W. Rao and P. W. H. Chan, J. Org. Chem., 2011, 76, 2521–2531 CrossRef CAS.
- X. Zhang, W. T. Teo, S. Sally and P. W. H. Chan, J. Org. Chem., 2010, 75, 6290–6293 CrossRef CAS.
- W. Huang, P. Zheng, Z. Zhang, R. Liu, Z. Chen and X. Zhou, J. Org. Chem., 2008, 73, 6845–6848 CrossRef CAS.
- G. Li, G. Zhang and L. Zhang, J. Am. Chem. Soc., 2008, 130, 3740–3741 CrossRef CAS.
- J. Muzart, Eur. J. Org. Chem., 2007, 2007, 3077–3089 CrossRef.
- H. Qin, N. Yamagiwa, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2007, 46, 409–413 CrossRef CAS.
- W. Rao, X. Zhang, E. M. L. Sze and P. W. H. Chan, J. Org. Chem., 2009, 74, 1740–1743 CrossRef CAS.
- Y. Tamaru, Eur. J. Org. Chem., 2005, 2005, 2647–2656 CrossRef.
- S. Wang, Y. Zhu, Y. Wang and P. Lu, Org. Lett., 2009, 11, 2615–2618 CrossRef CAS.
- M. Yoshimatsu, T. Otani, S. Matsuda, T. Yamamoto and A. Sawa, Org. Lett., 2008, 10, 4251–4254 CrossRef CAS.
- M. Yoshimatsu, T. Yamamoto, A. Sawa, T. Kato, G. Tanabe and O. Muraoka, Org. Lett., 2009, 11, 2952 CrossRef CAS.
- Z.-P. Zhan, X.-B. Cai, S.-P. Wang, J.-L. Yu, H.-J. Liu and Y.-Y. Cui, J. Org. Chem., 2007, 72, 9838–9841 CrossRef CAS.
- X. Zhang, W. T. Teo and P. W. H. Chan, Org. Lett., 2009, 11, 4990–4993 CrossRef CAS.
- R. Sanz, D. Miguel, A. Martinez, J. M. Alvarez-Gutierrez and F. Rodriguez, Org. Lett., 2007, 9, 727 CrossRef CAS.
- G. A. Olah, J. Am. Chem. Soc., 1972, 94, 808–820 CrossRef CAS.
- C. Poriel, F. Barrière, D. Thirion and J. Rault-Berthelot, Chem.-Eur.J., 2009, 15, 13304–13307 CAS.
- P. D. Mayo and H. Takeshita, Can. J. Chem., 1963, 41, 440–449 CrossRef.
- W. S. Rapson and R. Robinson, J. Chem. Soc., 1935, 1285–1288 RSC.
- M. Capponi, I. Gut and J. Wirz, Angew. Chem., 1986, 98, 358–359 CrossRef CAS.
- H. Mori, T. Matsuo, Y. Yoshioka and S. Katsumura, J. Org. Chem., 2006, 71, 9004–9012 CrossRef CAS.
- H. Sajiki, A. Mori, T. Mizusaki, T. Ikawa, T. Maegawa and K. Hirota, Org. Lett., 2006, 8, 987–990 CrossRef CAS.
- H. Kotsuki, P. K. Datta, H. Hayakawa and H. Suenaga, Synthesis, 1995, 1995, 1348–1350 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NOESY NMR spectra and X-ray crystal structure of compound 3a. CCDC reference numbers 879659. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra20852k |
| This journal is © The Royal Society of Chemistry 2012 |