Dielectric relaxation spectroscopy for the binary system of 1-butyl-3-methylimidazolium hexafluorophosphate and ethanol: interactions and micro phase behavior
Liyan
Ma
and
Kongshuang
Zhao
*
College of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: zhaoks@bnu.edu.cn
First published on 11th September 2012
Abstract
The interaction between the ionic liquid (IL) 1-butyl-3-methylimidazolium hexafluorophosphate ([bmim][PF6]) and ethanol and the micro phase behavior for the binary system have been investigated by dielectric relaxation spectroscopy (DRS) over a frequency range from 10 MHz to 20 GHz under constant temperature. The dielectric spectra with two relaxation processes were observed, and it could be satisfactorily fitted by an empirical equation including a non-Debye process at lower frequencies and a Debye process at higher frequencies. Detailed analysis indicates that either of the two relaxation processes contain the contributions of different polarization mechanisms, respectively. The low-frequency process, centered at about 1 GH is mainly due to the reorientation of the polar cation [bmim]+ and ion pairs formed between cation and anion of the IL and cooperative dynamics of the ethanol H-bond system; the high-frequency one, located at about 10 GHz is caused by the fast fluctuation of ion-pairs formed between cation and anion of the IL within the inter-ion distance and the rotation of singly H-bonded ethanol monomers at the ends of the ethanol chain. The ethanol concentration dependency of all the relaxation parameters shows a inflection point at the ethanol mass fraction of 20%, which is consistent with macro phase diagram. This inflection point is the cut-off point of one phase and two phase. The dielectric analysis indicates that a great micro-structure difference exists above and below 20%.
1 Introduction
In the last few years, ionic liquids (ILs) have attracted great attention because they are expected to have potential as green solvents in a variety of areas1–6 such as synthesis, catalysis, electrochemistry, separations and so on. Meanwhile, the increasing utilization of ionic liquids undoubtedly requires some systematic data of thermodynamic and thermophysical properties, especially solubility and transport properties, like electric conductivity and mutual diffusivity and so on. To aid in the development of ionic liquids for reactions and separations, the liquid phase equilibria of ionic liquids have been studied in a variety of different ways, such as IR, UV and quantum chemical calculations.7 The data obtained by these methods include binary temperature composition curves of ILs with alcohols,8–13 alkanes and aromatics,14 and water,9,15–17 ternary temperature composition curves of ILs with alcohols and water.9,18 Although there is a general understanding of the liquid phase behavior of ionic liquids with some organics, the information on the micro-structure of the mixture is still inadequate, which limits the further application of ionic liquid. Therefore, it is necessary to make a deeper analysis of the micro-structure and interaction to understand the relation between the micro-structure and macroscopic phenomenon.In recent years, the synthesis, properties and applications of a variety of ionic liquids have been studied, but relatively few of these investigations have focused on the basic physicochemical properties of their mixtures with cosolvents, especially with regards to micro-structure and interaction. For the microscopic description of the structure, Dupont has made a rough model,19,20 according to which, pure imidazolium-based ILs form an extended hydrogen-bond network composed of the cation and anion of the IL and when a polar cosolvent is added to the pure IL, this network breaks up, forming some aggregates. With the increase in cosolvent concentration, the aggregates are broken down into triple ions (TIs), contact ion pairs (CIPs), solvent-shared ion pairs (SIPs), solvent-separated ion pairs (S-SIPs) and free ions in turn as described in the literature.19 For these species of Dupont's scheme, many groups have tried to verify by various means as follows. Wang et al.21 studied the microscopic structures of 1-butyl-3-methylimidazolium tetrafluoroborate ([bmim][BF4]) and acetonitrile (CH3CN) mixtures by molecular dynamics simulation, their results showed that the interactions between the cations and anions and between the CH3CN molecules were enhanced after mixing. Katsuta et al.22,23 determined the ion pair formation constants of a series of ILs in dichloromethane and water by the conductance method, respectively. Bowers et al.24 investigated the aggregation behavior of three ionic liquids in aqueous solutions by means of surface tension, conductivity, and small-angle neutron scattering (SANS) measurements, from analysis of the SANS data, they proposed that models for the shapes and sizes of the aggregates were different according to the length of the carbon chain of the imidazolium cation and the types of anion. In addition, Wang25–27 and Zheng et al.28 also investigated the aggregation behavior of IL in salt solutions by means of surface tension, conductivity, fluorescence detection and other methods. The aggregation behavior of IL in aqueous or in organic solutions and the micro-structure of mixtures can be ascribed to various interactions between the constituent ions of IL and solvents.
From the above analysis, it is explicit that there have been many available techniques to study the structure and dynamics of ILs and their mixtures. Nevertheless, they only obtain macro information of whole solutions to infer inner structure without directly indicating micro behavior. On the other hand, NMR, Raman and other spectroscopic techniques generally provide information only about short-range (bonding) interactions and have a specific weakness with regard to the detection of S-SIPs. Dielectric relaxation spectroscopy (DRS) has obvious advantages to solve these problems because it is sensitive to the polarization processes related to molecular-level dipoles, including those associated with fluctuations of the ion pairs inside the sample. In addition, most of the cations and anions of ILs and the ion pairs composed of these ions have a dipole moment, this shows that studying ILs and their mixtures with molecular solvents by DRS is particularly appropriate and a preferential choice. Therefore a number of studies on dielectric properties of ILs have been reported.29–31 For example, Nakamura et al.32 investigated systematically the dynamic behaviors of ILs consisting of a series of 1-alkyl-3-methylimidazolium cations and various anionic species over a frequency range from 1 MHz to 20 GHz at room temperature using DRS and analyzed the relaxation mechanism with model schemes. Richert33 and Sangoro34et al. investigated charge transport, dipolar relaxations and the dynamics of solvation of the glass-forming ionic liquid. Weingartner et al.35–38 measured the static dielectric permittivity of a variety of ionic liquids and explained the essential difference among them. However, so far few studies have focused on the micro structure of IL/cosolvent mixtures by DRS. In recent years, Steinhauser et al.39–41 simulated some dielectric spectra of IL/co-solvent mixtures. Buchner et al. also started in detail to study the dielectric properties of many IL/cosolvent mixtures such as the binary mixtures of [bmim][BF4] and acetonitrile, methanol, dimethylsulfoxide (DMSO), dichloromethane (DCM),42 water, respectively, 1-ethyl-3-methylimidazolium ethylsulfate and dichloromethane,43N-methyl-N-ethylpyrrolidinium dicyanamide and dichloromethane.44 Their results showed that only a kind of ion-pair species formed in water41 and DMSO45 for concentrations of [bmim][BF4] C[bmim][BF4] < 3.5 M. For acetonitrile this limitation of concentration was at about 2.5 M. With the IL mixture continuously diluted, the cooperative dynamics typical for ILs transitioned to that of “normal” electrolyte solutions. The transition concentration varies depending on different solvents.
Undoubtedly, the research above revealed the relationship between bulk macro properties and micro-structure in the binary systems of ILs from a microscopic point of view to some extent. However, the microscopic model of interaction is still not clear, especially about the research on ILs with organic solvents. In addition, in practice, the mixture of ILs with alcohols is often used to separate or adjust the properties of the IL, and the intermiscibility of the IL with alcohols becomes more remarkable. As an example, the study on the liquid equilibria of [bmim][PF6] and ethanol has been reported in the literature.3 However, these studies were only limited to the macroscopic description of the phase diagram, and did not refer to the nature of the macroscopic phase behavior. In other words, a deeper analysis of micro-structure and interactions of the mixture of [bmim][PF6] and ethanol by DRS have not been reported so far to our knowledge.
In this paper, the dielectric spectra of a series of mixtures composed of [bmim][PF6] and ethanol have been analyzed in detail to investigate the interplay of ethanol and [bmim][PF6] in the mixtures. The micro-structure of these mixed solutions was analyzed theoretically from the view of the interaction and polarization. Furthermore, the macroscopic phase behavior was explained based on internal structure and the dynamics of binary mixtures.
2 Materials and method
2.1 Materials
The IL [bmim][PF6] (purity > 99.2%) used in this work was purchased from Shanghai Cheng Jie Chemical Co. LTD. China. The residual chloride in this product is less than 800 ppm, and water content is less than 1000 ppm. Anhydrous alcohol was purchased from Beijing Chemical Works, China. A series of binary mixtures composed of IL and ethanol with following ethanol mass fraction Wet were prepared: 0%, 5%, 10%, 15%, 20%, 40%, 60%, 80%, 85%, 90%, 100%. At Wet < 20%, the mixture was transparent liquid, and within Wet > 20%, the system showed turbid.2.2 Dielectric measurement
The dielectric spectra were measured with an Agilent E8362B PNA series network analyzer (Agilent Technologies, made in America), equipped with an Agilent 85070E open-ended coaxial probe (Agilent Technologies, made in America), which covers the frequency ranges from 10 MHz to 20 GHz. The permittivity and total dielectric loss were automatically calculated as functions of frequency by the built-in software of this measuring system. The system was tested and calibrated in accordance with the procedures recommended by the manufacturers. All measurements were conducted at 21 ± 0.1 °C. Because the dielectric measurement was completed in seconds, being an extremely fast process, the environment around the probe could be considered the same as the subject environment during uniform turbidity before the emergence of a macroscopic two-phase system.2.3 Dielectric data analysis
In an applied electric field of frequency f, the dielectric properties of samples can be characterized by the complex permittivity ε*, which is defined as:46| ε* = ε − jε′′ | (1) |
Where ε and ε′′ are the permittivity and the dielectric loss, respectively. The measured dielectric loss η′′ consists of two parts, one part is the contribution of direct current (dc) electric conductivity ε′′dc (κl/ε0ω) (κl, ε0, ω = (2πf) are the low-frequency limit of conductivity, the permittivity of vacuum and angular frequency, respectively), and another is the dielectric loss ε′′.ε′′dc can not be neglected for high conductivity systems including the case of this work. Thus κl was calculated from the scaling dependence of total dielectric loss with a slope −1 on ω according to 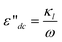 .47
.47
Therefore, the dielectric loss ε′′ of eqn (1) can be corrected as:
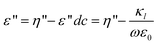 | (2) |
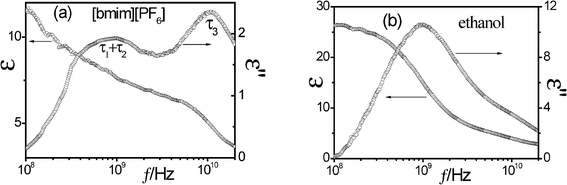
Dielectric relaxations can be characterized by a set of dielectric relaxation parameters (or dielectric parameters): εl, εh, or Δε = (εl − εh), τ = 1/(2πf0) (f0 is the relaxation frequency) and β (0 < β ≤ 1), they are the low-frequency and high-frequency limits of permittivity, dielectric increment, relaxation time and distribution parameter of relaxation time, respectively. All these dielectric parameters can be obtained by fitting the following eqn (3) including one (i = 1) or two (i = 1, 2) Cole-Cole's term48 to experimental and calculated dielectric spectra:
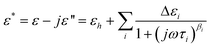 | (3) |
3 Results and discussion
3.1 Dielectric spectra for the mixture of [bmim][PF6] (IL) and ethanol
Fig. 1 shows the three-dimensional representations of the experimental results: it is the frequency dependence of dielectric loss of the binary system of [bmim][PF6] and ethanol. From Fig. 1, two relaxations can be observed at about 109 HZ and 1010 HZ, respectively. The low-frequency relaxation first decreases and then increases as ethanol concentration (mass fraction) increases, a turning point appears at the ethanol mass fraction of 20%. The relaxation frequency of high frequency changes a little, but it is around 20%. In addition, whether low- or high-frequency relaxation has a certain distribution, it means that the relaxation may contain sub-relaxation.![3-D representation for the frequency dependency of dielectric loss of the binary mixtures of [bmim][PF6] and ethanol with different ethanol mass fraction.](/image/article/2012/RA/c2ra20732j/c2ra20732j-f1.gif) | ||
| Fig. 1 3-D representation for the frequency dependency of dielectric loss of the binary mixtures of [bmim][PF6] and ethanol with different ethanol mass fraction. | ||
3.2 Determination of relaxation parameters
To examine the unique dielectric behavior in detail and verify possible relaxation mechanisms, the Cole-Cole equation, i.e., eqn (3) (where i = 2), was used to fit the dielectric spectra. Several dielectric parameters that reflect the relaxation processes in whole measuring frequency were obtained and are listed in Table 1. For clarity, the dielectric increments, Δεl and Δεh and relaxation times, τl and τh of low- and high-frequency relaxation processes were plotted as a function of ethanol mass fraction Wet in Fig. 2, respectively.![Low-, high-frequency dielectric increment (a) and low-, high-frequency relaxation time (b) as a function of ethanol mass fraction in the mixtures of [bmim][PF6] and ethanol at 21 °C.](/image/article/2012/RA/c2ra20732j/c2ra20732j-f2.gif) | ||
| Fig. 2 Low-, high-frequency dielectric increment (a) and low-, high-frequency relaxation time (b) as a function of ethanol mass fraction in the mixtures of [bmim][PF6] and ethanol at 21 °C. | ||
| w%−ethanol | x −ethanol | Δεl | β | Δεl−IL | Δεl−ethanol | Δεh | Δεh−IL | Δεh−ethanol | τ l /ps | τ h /ps |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 0 | 2.57 | 0.52 | 2.57 | 0 | 3.23 | 3.23 | 0 | 159 | 13.9 |
| 5 | 0.25 | 4.68 | 0.54 | 1.94 | 6.26 | 3.74 | 2.44 | 0.70 | 190 | 15.4 |
| 10 | 0.41 | 3.68 | 0.55 | 1.52 | 10.4 | 3.94 | 1.92 | 1.16 | 210 | 16.6 |
| 15 | 0.52 | 5.98 | 0.58 | 1.23 | 13.3 | 4.14 | 1.55 | 1.49 | 253 | 16.8 |
| 20 | 0.61 | 3.16 | 0.60 | 1.01 | 15.5 | 4.61 | 1.27 | 1.73 | 290 | 17.9 |
| 40 | 0.80 | 12.6 | 0.78 | 0.50 | 20.5 | 4.03 | 0.63 | 2.30 | 257 | 16.4 |
| 60 | 0.90 | 13.5 | 0.85 | 0.25 | 23.0 | 3.67 | 0.32 | 2.58 | 238 | 15.7 |
| 80 | 0.96 | 19.7 | 0.88 | 0.10 | 24.5 | 3.27 | 0.13 | 2.75 | 180 | 15.4 |
| 85 | 0.97 | 20.3 | 0.90 | 0.07 | 24.8 | 3.15 | 0.09 | 2.78 | 174 | 15.5 |
| 90 | 0.98 | 22.4 | 0.93 | 0.05 | 25.1 | 3.02 | 0.06 | 2.81 | 170 | 15.5 |
| 100 | 1.00 | 25.5 | 0.95 | 0 | 25.5 | 2.86 | 0 | 2.86 | 156 | 15.4 |
From Fig. 2, a break at Wet = 20% can be observed for all the relaxation parameters, which divide the Wet into two regions. This shows that the relaxation behavior existing in the two regions are dominated by different relaxation mechanisms. The inflection point at 20% is worth paying attention to because it is closely related to the macro phase separation according to the literature9 in which phase separation occurs exactly at Wet = 20%, when ethanol mass fraction exceeds 20%, the macro phase behavior of the mixture transformed from transparent liquid to turbid liquid. This shows that the mechanism of interaction between ethanol and ionic liquid changed dramatically when the proportion of ethanol molecule in the mixtures reached a critical value. This article aims to understand the essence of the phase behavior by analyzing the relaxation mechanism.
3.3 Relaxation mechanism
 | ||
| Fig. 3 Dielectric permittivity, ε(f), and dielectric loss, ε′′(f), spectra of pure IL (a) and ethanol (b). | ||
For the IL [bmim][PF6] in Fig. 3(a), according to the literature,49–51 the low-frequency relaxation may be mainly ascribed to the reorientation of the dipolar cations, and partly the rotation polarization of ion pairs, which correspond to the horizontal dipole moment for τ1 and vertical dipole moment for τ2 in Fig. 4, respectively. In other words, the low-frequency relaxation of the IL is caused by the sum of contributions of reorientation polarization of the cations and rotation polarization of ion pairs. Similarly, according to the literature,32 the high-frequency relaxation can be considered to be caused by the inter-ion motion between cation [bmim]+ and anion [PF6]− of formed ion-pairs, which correspond to τ3 in Fig. 4. For ethanol of Fig. 3(b), the low-frequency relaxation, corresponding to the non-Debye mode, can be ascribed to the cooperative dynamics of the H-bond system, while the high-frequency Debye mode can be assigned to the rotation of singly H-bonded ethanol monomers at the ends of the ethanol chain.52,53
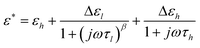 | (4) |
![Schematic representation of the relaxation mechanisms for IL [bmim][PF6]. τ1, τ2 and τ3 indicate the relaxation time corresponding to different polarization mode, respectively.](/image/article/2012/RA/c2ra20732j/c2ra20732j-f4.gif) | ||
| Fig. 4 Schematic representation of the relaxation mechanisms for IL [bmim][PF6]. τ1, τ2 and τ3 indicate the relaxation time corresponding to different polarization mode, respectively. | ||
![Dielectric loss, ε′′(ƒ), spectrum of mixture of [bmim][PF6] and ethanol (Wet = 85%) . Open circles represent experimental data, full line is best-fit curve calculated from the eqn (4), shaded areas represent the contributions of the individual processes.](/image/article/2012/RA/c2ra20732j/c2ra20732j-f5.gif) | ||
| Fig. 5 Dielectric loss, ε′′(ƒ), spectrum of mixture of [bmim][PF6] and ethanol (Wet = 85%) . Open circles represent experimental data, full line is best-fit curve calculated from the eqn (4), shaded areas represent the contributions of the individual processes. | ||
Lower frequency non-Debye processes. As shown in Fig. 5, the low-frequency mode in the DR spectra of [bmim][PF6] and ethanol mixtures, centered at about 1 GHz, was satisfactorily modeled by a Cole-Cole empirical equation which means a distribution of relaxation times. To distinguish the contribution of constituent in the mixture to the relaxation mode, this mode was subdivided in terms of mixture composition. On the basis of relaxation mechanism of pure constituent discussed above, the relaxation mechanism of the binary mixtures can be considered to come from the contributions of the reorientation of the dipolar [bmim]+ and ion pairs and cooperative dynamics of the H-bond system, which can be explained by analyzing the variation trend of dielectric parameters in Fig. 2(a) and (b). For the dielectric increment of the low-frequency relaxation Δεl, in the Wet ≤ 20% range, the values of Δεl are almost unchanged while they increase gradually at Wet > 20%. This is because when the ethanol content is very low (Wet ≤ 20%), large ionic liquid super-molecular structure interspersed with a few ethanol molecules by hydrogen bonding, which enhanced the stability of orientation of [bmim]+ and ion-pair and ethanol H-bond. As a result, Δεl remains unchanged, meanwhile, it will take longer time to complete these orientation polarization processes, i.e. τl increases. As the ethanol content increases and the weight ratio exceeds 20%, ethanol molecule itself formed a network structure by hydrogen bonding, forming ethanol zone, and IL super-molecular structures formed gradually again, generating IL zone, making the orientation easier and the value of Δεl increases gradually, accordingly the relaxation time τl needed for orientation polarization shortens gradually.
On the other hand, various theories have been applied to connect the observed relaxation behavior with molecular-level processes. Among them, the Cavell equation54 relates the dielectric increment Δεl to the effective dipole moment μeff,i of the species responsible for that process:
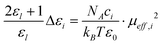 | (5) |
Where NA and kB are the Avogadro constant and Boltzmann constant, respectively, T is thermodynamic temperature, and ci the molar concentration of the species i in mixtures. In fact, the low- or high-frequency dielectric increment Δεl or Δεh (seen in Table 1) consists of the contributions of IL and ethanol, each of which depends on (proportional to) the concentration of each constituents in the mixed system. Therefore, Δεl can be approximated by the following formula:
| Δεl ≈ Δεlapp = Δεl−ethanol + Δεl−IL ≅ xethanolΔεethanol + xILΔεIL | (6) |
That is,
| Δεl−ethanol = xethanolΔεethanol | (7a) |
| Δεl−IL = xILΔεIL | (7b) |
Where xethanol and xIL are the molar fraction of ethanol and IL, respectively, Δεethanol and ΔεIL are the dielectric increment of pure ethanol and IL, respectively. It should be noted that Δεlapp calculated from the sum of Δεl−ethanol and Δεl−IL is an approximate value, which ignores the interaction between IL and ethanol, therefore, it is greater than that of experimental Δεl (seen in Table 1). Representing the contributing degrees of Δεethanol and ΔεIL to the low-frequency relaxation Δεl by molar fraction of ethanol and IL in the mixture, as weight factor, is approximate. Nevertheless, it may reflect the real situation because the relaxation frequency for pure ethanol or pure IL is very close to that in mixture, respectively. Thus, effective dipole moment μl,i of various relaxation species of low-frequency were obtained by the calculated Δεl−ethanol and Δεl−IL into eqn (5) and the results are listed in Table 2.
| w%−ethanol | μ l−ethanol/D | μ l−IL/D | μ h−ethanol/D | μ h−IL/D | κ l /(s m−1) |
|---|---|---|---|---|---|
| 0 | 2.44 | 2.74 | 0.19 | ||
| 5 | 6.95 | 2.20 | 2.33 | 2.47 | 0.41 |
| 10 | 6.45 | 2.05 | 2.16 | 2.30 | 0.37 |
| 15 | 6.04 | 1.92 | 2.02 | 2.15 | 0.87 |
| 20 | 5.74 | 1.82 | 1.92 | 2.04 | 0.88 |
| 40 | 4.94 | 1.57 | 1.65 | 1.76 | 0.65 |
| 60 | 4.51 | 1.43 | 1.51 | 1.61 | 0.47 |
| 80 | 4.22 | 1.34 | 1.41 | 1.50 | 0.21 |
| 85 | 4.17 | 1.32 | 1.39 | 1.48 | 0.15 |
| 90 | 4.12 | 1.31 | 1.38 | 1.46 | 0.14 |
| 100 | 4.03 | 1.35 | 0.01 |
For clarity, the values of μl,i were plotted as a function of ethanol mass fraction Wet in Fig. 6. As can be seen from Fig. 6 both μl,i decreases gradually with increasing Wet, while the slope of decrease differs above and below 20%. For IL, μl−IL is near 2 D, which is a smaller value to that of the literature, μ ≈ 5 D42 indicating that IL network structures were tighter due to the interaction between ethanol and IL. For ethanol, effective dipole moment μl,ethanol decreases gradually from about 7 D to 4 D with increasing Wet (see Table 2), in the Wet ≤ 20% range, the ethanol is mainly surrounded by network structure of ionic liquids, and the distance between electropositivity and electronegativity produced by hydrogen bond decreases with increasing Wet, accordingly, μl,ethanol decreases gradually. At Wet ≥ 20%, ethanol itself quickly formed a hydrogen bond system, and μl,ethanol is almost only influenced by ethanol, thus, μl,ethanol also decreases slowly as Wet increases. The amplitude of decrease above and below 20% is related to the strength of both interactions: in the Wet ≤ 20% range, the interaction arises between the IL and ethanol, which has a bigger effect on either IL or ethanol, resulting in more steep slope, instead, at Wet ≥ 20%, either IL or ethanol is influenced by itself, therefore, the slope becomes less steep.
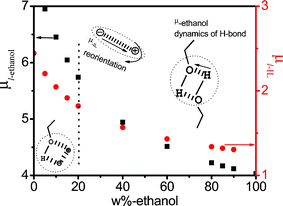 | ||
| Fig. 6 The dependence of micro-dipole moment μl of various relaxation species of low-frequency on the concentration of ethanol. | ||
Higher frequency Debye processes. The high-frequency mode in the DR spectra of [bmim][PF6] and ethanol mixtures, centered at about 10 GHz, was well modeled by a Debye type relaxation as shown in Fig. 5. This relaxation can be considered to be from the contributions of two independent micro-polarization modes, namely, fast fluctuation of ion-pairs formed by cationic and anionic of the IL within the inter-ion distance and the rotation of singly H-bonded ethanol monomers at the ends of the ethanol chain. From Fig. 2(a), it is obvious that the variation trend of dielectric increment of high-frequency relaxation Δεh with Wet is totally different from that of low-frequency, showing radically different polarization processes between the two relaxations. Considering the case of pure constituent of the binary mixtures discussed in section 3.3.1, that's understandable. For the Δεh, at Wet below 20%, it increases monotonously while decreases monotonously above 20%. This may be interpreted as follows: at Wet ≤ 20%, the interaction between IL and ethanol molecules weakened the stability of the inter-ion distance of [bmim][PF6] and ethanol monomers, resulting in linear increase of Δεh. While at Wet ≥ 20%, as Wet increases, in ethanol zone and IL zone, the stability of the relaxation species increases gradually, thus, Δεh decreases gradually. In contrast, the dependence of the relaxation time of the high-frequency relaxation τh on the Wet shows a very similar variation trend with that of low-frequency relaxation (see Fig. 2(b)). At Wet ≤ 20%, the interaction between IL and ethanol results in the fluctuation of the inter-ion distance and rotation of singly H-bonded ethanol monomers were not easy to catch up with electric field change, i.e. the polarization caused by them was easier to occur, leading to the increase of τh gradually. At Wet above 20%, the interaction between ethanol and IL was weakened, no matter in which zone, the relaxation species was affected less, so they were more free and easier to keep up with the electric field change, leading to a sharp decrease of τh. As ethanol content continues to rise, the two structures stabilize quickly and τh levels off.
Similarly, the high-frequency dielectric increment Δεh can also be represented approximately by the contributions of pure ethanol and IL:
| Δεh = Δεh−ethanol + Δεh−IL ≅ xethanolΔεethanol + xILΔεIL | (8) |
Its weight concentration, xethanol and xIL, reflects the contributing degrees to the high-frequency relaxation. It should be noted that, current Δεethanol and ΔεIL are the high-frequency dielectric increments of pure ethanol and IL, respectively. Using the Δεh−ethanol, Δεh−IL calculated above and eqn (5), the effective dipole moment μh,i of various relaxation species of high-frequency was also calculated and the results are listed in Table 2.
For clarity, the values of μh,i were plotted as a function of ethanol mass fraction Wet in Fig. 7. As can be seen in Fig. 7 that the variation trends of μh,ethanol and μh,IL are basically same as that of low-frequency effective dipole moments μl,i For IL, μh,IL is almost consistent with μl−IL, indicating that the effective dipole moments of low- and high-frequency are mainly related to the IL super-molecular network. While for ethanol, μh,ethanol is smaller than μl−ethanol, suggesting that the effective dipole moment caused by cooperative dynamics of the H-bond system is larger than that derived from the rotation of ethanol monomers. The decrease trend in less and more than 20% can be explained as follows: in Wet ≤ 20% range, the inter-ion distance motion of ion-pairs and rotation of ethanol monomers both existed in the network structure formed by the interaction between IL and ethanol, thence, the decrease magnitude is relatively larger, on the contrary, at Wet ≥ 20%, IL and ethanol are only affected by themselves, consequently, the trend decreases more slowly.
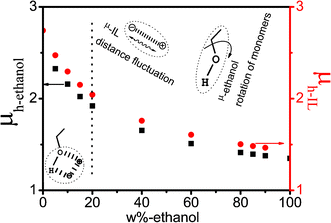 | ||
| Fig. 7 The dependence of micro-dipole moment μh of various relaxation species of high-frequency on the concentration of ethanol. | ||
3.4 Micro-interpretation of macro-phase behavior
From the above analysis result, the ethanol content dependence of all dielectric parameters shows a turning point at the ethanol mass fraction of 20%. The turning point coincides exactly with the macro phase diagram reported in the literature,9,16,18 and the point of 20% ethanol content divides the phase diagram into a single-phase region and double-phase region as shown in Fig. 8. This means that macroscopic phase separation is closely related to the micro-structural change of the binary mixture system, essentially a result of the interaction between ethanol and ionic liquids.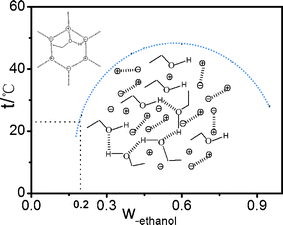 | ||
Fig. 8 The structure schematic drawings of the interaction between ethanol and ionic liquids.  , ,  represent cationic and anionic of IL, respectively. represent cationic and anionic of IL, respectively. | ||
Because there exist different interactions in the binary mixture system composed of ethanol and [bmim][PF6]: the electrostatic interaction between the electronegative oxygen atoms in ethanol and the cation [bmim]+, the interaction between the oxygen atoms and C2–H of [bmim]+ by hydrogen bonds, and weak interaction between the hydrogen atom of hydroxyl in ethanol and [PF6]− as shown in Fig. 8, in the measuring temperature of 21 °C, according to the phase diagram, at Wet ≤ 20%, the system is in one phase region in which large ionic liquid super-molecular structure interspersed with a few ethanol molecules by various interactions, phase behavior of the mixture was transparent one phase liquid in macro sense, while in Wet ≥ 20%, the mixture became turbid, with increasing Wet, ethanol molecule itself also formed the hydrogen bond structure as shown in the graphical representation Fig. 8. The system contains the properties of both IL and ethanol molecules which shows turbid two phase liquid in macro sense.
3.5 Analysis of conductivity κl
Fig. 9 shows the plot of the dc-conductivity κl listed in Table 2 against the ethanol mass fraction Wet. In this binary mixture system of [bmim][PF6] and ethanol, the value of κl is proportional to the density of charge carriers and inversely proportional to medium viscosity. In the Wet ≤ 20% range, as the Wet increases, although the concentration of the ions that make electrical conducting electricity decreases, the viscosity of the mixture decreases quickly, leading to the ion mobility increasing, as a result, κl increases. At Wet ≥ 20%, with Wet, ethanol itself formed up chain-like hydrogen bonding structure, the concentration of positive and negative ions of IL decreases, in addition, the association of the anion and cation reduces the concentration of conductive particles, therefore κl declines gradually to almost zero.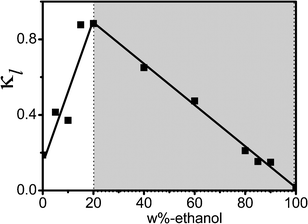 | ||
| Fig. 9 The dependence of dc-conductivity κl on the concentration of ethanol. | ||
4 Conclusion
Dielectric spectra of some mixtures containing ionic liquid [bmim][PF6] and ethanol with varying ethanol concentration were studied systematically over a frequency range from 10 MHz to 20 GHz under constant temperature. Two relaxations were found at about 109 HZ and 1010 HZ, respectively. Several dielectric parameters that reflect the relaxation processes were obtained by using the Cole-Cole equation to fit the dielectric data. The micro-relaxation mechanisms were well interpreted through analysis of these parameters. It was concluded that the micro-structure of the binary system and the strength of interactions in the system exist distinct diversity in two regions depending on ethanol content. On this basis, the macro phase behavior above and below 20% ethanol content was also well explained.The present study also shows that the results of the dielectric analysis of the binary system composed of [bmim][PF6] and ethanol support Dupont's model commendably. There are mainly IL super-molecular structures in IL-rich region. At Wet ≤ 20%, large ionic liquid super-molecular structure interspersed with a few ethanol molecules by various interactions, the system shows transparent one phase liquid in macro sense. At Wet ≥ 20%, with increasing Wet, the interaction between IL and ethanol weakened, IL zone consists of the IL super-molecular structure, meanwhile, ethanol molecules formed chain-like hydrogen bonding structure with only a spot of CIP and SIP around, triggering ethanol zone. As a result, a turbid two phase system emerges. These results provide some theoretical references for further applications in the separation of ionic liquids, and also prove DRS is appropriate for studying the microstructure of the binary system of IL.
Acknowledgements
The authors wish to thank Ms. J. M. Pan and Dr S. J. Zhao for the provision of laboratory facilities for high-frequency dielectric measurements. Financial support of this work by the National Natural Science Foundation of China (Grants 21173025 and 20976015) is gratefully acknowledged.References
- X. X. Han and D. W. Armstrong, Acc. Chem. Res., 2007, 40, 1079–1086 CrossRef CAS PubMed.
- T. L. Greaves and C. J. Drummond, Chem. Soc. Rev., 2008, 37, 1709–1726 RSC.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2008, 108, 206–237 CrossRef CAS PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- A. Pinkert, K. N. Marsh, S. S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712–6728 CrossRef CAS PubMed.
- F. V. Rantwijk and R. A. Sheldon, Chem. Rev., 2007, 107, 2757–2785 CrossRef PubMed.
- K. N. Marsh, J. A. Boxall and R. Lichtenthaler, Fluid Phase Equilib., 2004, 219, 93–98 CrossRef CAS.
- C. T. Wu, K. N. Marsh, A. V. Deev and J. A. Boxall, J. Chem. Eng. Data, 2003, 48, 486–491 CrossRef CAS.
- V. Najdanovic-Visak, J. Esperanca, L. P. N. Rebelo, M. N. da Ponte, H. J. R. Guedes, K. R. Seddon and J. Szydlowski, Phys. Chem. Chem. Phys., 2002, 4, 1701–1703 RSC.
- U. Domanska, E. Bogel-Lukasik and R. Bogel-Lukasik, J. Phys. Chem. B, 2003, 107, 1858–1863 CrossRef CAS.
- M. Wagner, O. Stanga and W. Schroer, Phys. Chem. Chem. Phys., 2003, 5, 3943–3950 RSC.
- J. M. Crosthwaite, N. V. Sudhir, K. Aki, E. J. Maginn and J. F. Brennecke, J. Phys. Chem. B, 2004, 108, 5113–5119 CrossRef CAS.
- J. M. Crosthwaite, M. J. Muldoon, N. V. Sudhir, K. Aki, E. J. Maginn and J. F. Brennecke, J. Phys. Chem. B, 2006, 110, 9354–9361 CrossRef CAS PubMed.
- U. Domanska and A. Marciniak, J. Chem. Eng. Data, 2003, 48, 451–456 CrossRef CAS.
- J. E. L. Dullius, P. A. Z. Suarez, S. Einloft, R. F. de Souza, J. Dupont, J. Fischer and A. De Cian, Organometallics, 1998, 17, 815–819 CrossRef CAS.
- J. L. Anthony, E. J. Maginn and J. F. Brennecke, J. Phys. Chem. B, 2001, 105, 10942–10949 CrossRef CAS.
- M. H. Abraham, J. G. Huddleston and W. E. Acree, Ind. Eng. Chem. Res., 2003, 42, 413–418 CrossRef CAS.
- K. N. Marsh, A. Deer, A. C-T. Wu, E. Tran and A. Klamt, Korean J. Chem. Eng., 2002, 19(3), 357–362 CrossRef CAS.
- J. Dupont, J. Braz. Chem. Soc., 2004, 15, 341–350 CrossRef CAS.
- C. S. Consorti, D. H. Farrar and J. Dupont, J. Phys. Chem. B, 2005, 109, 4341–4349 CrossRef CAS PubMed.
- X. P. Wu, Z. P. Liu, S. P. Huang and W. C. Wang, Phys. Chem. Chem. Phys., 2005, 7, 2771–2779 RSC.
- S. Katsuta, K. Imai and H. Seki, J. Chem. Eng. Data, 2008, 53, 1528–1532 CrossRef CAS.
- S. Katsuta and R. Ogawa, J. Chem. Eng. Data, 2007, 52, 248–251 CrossRef CAS.
- J. Bowers, C. P. Butts and R. K. Heenan, Langmuir, 2004, 20, 2191–2198 CrossRef CAS PubMed.
- H. Y. Wang, Q. Q. Feng, J. J. Wang and H. C. Zhang, J. Phys. Chem. B, 2010, 114, 1380–1387 CrossRef CAS PubMed.
- H. Y. Wang, J. J. Wang, S. B. Zhang and X. P. Xuan, J. Phys. Chem. B, 2008, 112, 16682–16689 CrossRef CAS PubMed.
- J. J. Wang, H. Y. Wang, S. L. Zhang, H. C. Zhang and Y. Zhao, J. Phys. Chem. B, 2007, 111, 6181–6188 CrossRef CAS PubMed.
- B. Dong, N. Li, L. Q. Zheng, L. Yu and T. Inoue, Langmuir, 2007, 23, 4178–4182 CrossRef CAS PubMed.
- K. Yamamoto, M. Tani and M. Hangyo, J. Phys. Chem. B, 2007, 111, 4854–4859 CrossRef CAS PubMed.
- E. I. Izgorodina, M. Forsyth and D. R. MacFarlane, Phys. Chem. Chem. Phys., 2009, 11, 2452–2458 RSC.
- D. A. Turton, J. Hunger, G. Hefter, A. Thoman and R. Buchner, J. Am. Chem. Soc., 2009, 131, 11140–11146 CrossRef CAS PubMed.
- K. Nakamura and T. Shikata, ChemPhysChem, 2010, 11, 285–294 CrossRef CAS PubMed.
- N. Ito, W. Huang and R. Richert, J. Phys. Chem. B, 2006, 110, 4371–4377 CrossRef CAS PubMed.
- C. Krause, J. R. Sangoro, C. Iacob and F. Kremer, J. Phys. Chem. B, 2010, 114, 382–386 CrossRef CAS PubMed.
- C. Wakai, A. Oleinikova and M. O. H. Weingartner, J. Phys. Chem. B, 2005, 109, 17028–17030 CrossRef CAS PubMed.
- H. Weingartner, P. Sasisanker, C. Daguenet, P. J. Dyson, I. Krossing, A. Olleinikova, J. Slattery and T. Schubert, J. Phys. Chem. B, 2007, 111, 4775 CrossRef PubMed.
- C. Daguenet, I. Krossing, A. Oleinikova and H. Weingartner, J. Phys. Chem. B, 2006, 110, 12682–12688 CrossRef CAS PubMed.
- M. M. Huang, Y. P. Jiang, G. W. Driver and H. Weingartner, J. Chem. Eng. Data, 2011, 56, 1494–1499 CrossRef CAS.
- C. Schröder and O. Steinhauser, J. Chem. Phys., 2010, 132, 244109–244124 CrossRef PubMed.
- G. Neumayr, C. Schröder and O. Steinhauser, J. Chem. Phys., 2009, 131, 174509–174520 CrossRef CAS PubMed.
- C. Schröder, J. Hunger, A. Stoppa, R. Buchner and O. Steinhauser, J. Chem. Phys., 2008, 129, 184501 CrossRef PubMed.
- J. Hunger, A. Stoppa and R. Buchner, J. Phys. Chem. B, 2008, 112, 12913–12919 CrossRef CAS PubMed.
- J. Hunger, A. Stoppa, R. Buchner and G. Hefter, J. Phys. Chem. B, 2009, 113, 9527–9537 CrossRef CAS PubMed.
- S. Schrödle, G. Annat, D. R. MacFarlane, M. Forsyth, R. Buchner and G. Hefter, Chem. Commun., 2006, 1748–1750 RSC.
- G. Hefter, R. Buchner, J. Hunger and A. Stoppa, ACS Symposium Series, Washington, DC, 2010 Search PubMed.
- K. Asami, Prog. Polym. Sci., 2002, 27, 1617 CrossRef CAS.
- T. Mitsumata, J. P. Gong and K. I. Y. Osada, J. Phys. Chem. B, 1998, 102, 5246–5251 CrossRef CAS.
- K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341 CrossRef CAS.
- A. Stoppa, J. Hunger, R. Buchner, G. Hefter, A. Thoman and H. Helm, J. Phys. Chem. B, 2008, 112, 4854–4858 CrossRef CAS PubMed.
- M. Mizoshiri, T. Nagao, Y. Mizoguchi and M. Yao, J. Chem. Phys., 2010, 132, 164510 CrossRef PubMed.
- J. Hunger, A. Stoppa, S. Schrodle, G. Hefter and R. Buchner, ChemPhysChem, 2009, 10, 723–733 CrossRef CAS PubMed.
- T. Sato and R. Buchner, J. Phys. Chem. A, 2004, 108, 5007–5015 CrossRef CAS.
- P. Petong, R. Pottel and U. Kaatze, J. Phys. Chem. A, 2000, 104, 7420–7428 CrossRef CAS.
- E. A. S. Cavell, P. C. Knight and M. A. Sheikh, Trans. Faraday Soc., 1971, 67, 2225 RSC.
| This journal is © The Royal Society of Chemistry 2012 |