Strong reduced graphene oxide–polymer composites: hydrogels and wires†
Hongbin
Feng
ab,
Yueming
Li
b and
Jinghong
Li
*b
aDepartment of Chemistry, University of Science and Technology of China, Hefei 230026, China
bDepartment of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: jhli@mail.tsinghua.edu.cn; Fax: 86-10-62795290
First published on 6th July 2012
Abstract
By in situ chemical reduction of graphite oxide (GO) mixed with poly(vinyl alcohol) (PVA), we successfully fabricated reduced graphene oxide (rGO)–PVA composite hydrogels with improved dispersion and load transfer in their composites. The rGO–PVA composite wires were made by thermal-drawing or by directly drawing from the rGO–PVA dispersion and their mechanical properties were rapidly evaluated using a microfabricated tuning fork device. It was found that the Young's modulus of the polymer composites increase by ca. 200% with only 0.68 vol % addition of the rGO. The thermal properties of the composites were studied by differential scanning calorimetry (DSC), and it was observed that the addition of graphene to PVA greatly improves the thermal stability of the composites. Raman spectroscopy revealed the existence of an interaction between the graphene and the polymer via the shift in the vibration bands of the graphene in the composites.
Introduction
Graphene sheets like unrolled carbon nanotubes are unique nanomaterials having exceptional thermal, mechanical, and high electrical properties.1–5 Graphene sheets are thus attractive as atomically thin yet robust components for nanoelectronic devices and as nanoscale building blocks with a polymer matrix to fabricate graphene-based composites.6–10 Their mechanical strength is comparable to that of carbon nanotubes and their shapes may make them superior to nanotubes for some applications. In addition, recent studies suggested that the production cost of graphene sheets in large quantities could be much lower than that of carbon nanotubes, which might make them a cheaper replacement for nanotubes in some applications.11–17 Hence, it will be extremely interesting to fabricate composites with graphene sheets to improve their mechanical properties.18 In order to achieve high performance of the polymer composite, it is important not only to use the inherent properties of the nanofiller, but also to optimize the dispersion and improve interface load transfer. However, graphene sheets have a great tendency to spontaneously agglomerate due to strong van der Waals and π–π interactions. This problem has been encountered in all previous efforts aimed at the application and improvement of graphene-based composites materials.19 To solve this problems, GO was now prepared, which contains many oxygen functional groups, such as hydroxyls, epoxides, carboxyls and carbonyls, to improve the dispersion.Chemically converted graphene sheets obtained from polymer-treated GO sheets can readily form stable dispersions of individual sheets in organic solvents and water, making them more compatible with other materials, facilitating the preparation of composites. As the reduction proceeds, the presence of the polymer in solution was a key to preventing the agglomeration of the sheets, and the sheets become coated with the polymer and remained individually dispersed. Here, the way to improve the dispersion of a polymer blend with graphene is to incorporate specific chemical interactions between the components.
In this paper, rGO–PVA composite hydrogels were fabricated by in situ reduction of GO sheets mixed with PVA in N,N-dimethylformamide (DMF), showing good dispersion and load transfer in the polymer matrix. Composite wires were made by thermal-drawing or by directly drawing from the rGO–PVA dispersion. We have shown an increase of mechanical properties such as the Young's modulus of the composite wires, which increased by 200% while adding only 0.68 vol % of graphene to PVA, using a novel and simple microfabricated tuning fork device. Raman spectroscopy was used to prove the interaction between the graphene and the polymer via the shift in the vibration bands of the graphene in the composites. The DSC data exhibited the strong interaction between polymer matrix and nanofillers.
Experimental
Preparation of rGO–PVA composite hydrogels and wires
GO was prepared by the Hummers method35 from SP-1 graphite (Alfa aesar), and dried over phosphorus pentoxide in a vacuum desiccator for a week. Dried GO (50 mg) was dispersed in anhydrous DMF (5 mL), mixed with PVA (100 mg, from Aldrich, molecular weight range 124![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–186000 g mol−1, 99+% hydrolyzed) at 100 °C for 12 h, followed by homogenization and ultrasonication. Reduction of the dispersed materials was carried out with hydrazine (with 80% H2O, 0.1 mL in 10 mL of DMF, Sigma-Aldrich) at 80 °C for 24 h. Different quantities of the PVA matrix were added into the rGO–PVA dispersion and heating continued at 100 °C for 4 h. Upon completion, the coagulation of the polymer composites was accomplished by adding the DMF solutions dropwise into a large volume of vigorously stirred acetone (10:1 with respect to the volume of DMF used). The coagulated composite hydrogels were isolated via filtration; washed with acetone (200 mL), and dried at 110 °C under vacuum for 4 h to remove residual solvent, anti-solvent, and moisture. To convert wt % loading of rGO sheets in the composite samples to vol % (as used in the text), a density for the PVA-treated GO sheets of 2.2 g cm−1 was assumed. The composite wires were made by thermal-drawing or by directly drawing from the rGO–PVA dispersion.
000–186000 g mol−1, 99+% hydrolyzed) at 100 °C for 12 h, followed by homogenization and ultrasonication. Reduction of the dispersed materials was carried out with hydrazine (with 80% H2O, 0.1 mL in 10 mL of DMF, Sigma-Aldrich) at 80 °C for 24 h. Different quantities of the PVA matrix were added into the rGO–PVA dispersion and heating continued at 100 °C for 4 h. Upon completion, the coagulation of the polymer composites was accomplished by adding the DMF solutions dropwise into a large volume of vigorously stirred acetone (10:1 with respect to the volume of DMF used). The coagulated composite hydrogels were isolated via filtration; washed with acetone (200 mL), and dried at 110 °C under vacuum for 4 h to remove residual solvent, anti-solvent, and moisture. To convert wt % loading of rGO sheets in the composite samples to vol % (as used in the text), a density for the PVA-treated GO sheets of 2.2 g cm−1 was assumed. The composite wires were made by thermal-drawing or by directly drawing from the rGO–PVA dispersion.
Material characterization
The atomic-force microscopy (AFM) measurements were performed with a SPI 4000 system in tapping mode. The samples were deposited from ethanol suspensions onto freshly cleaved mica substrates, dried by evaporation, and the measurements were performed in air at ambient temperature and pressure. Electron micrographs were acquired using a field emission scanning electron microscope (SEM) (JSM 7401F) and a transmission electron microscope (TEM) (JEM-1200EX, JEOL, operated at 200 kV). SEM was used to characterize the dispersion of the graphene in the resultant composite samples. Optical microscopy images obtained by using an Olympus BX51 W/DP70 microscope were used to visualize the microscopic features of the composite wires. A Renishaw RM2000 microscopic confocal Raman spectrometer (Gloucestershire, United Kingdom) was used for Raman measurements with 633 nm excitation wavelengths at 25% energy and a laser spot size of 2.0 μm. Thermal properties of the composites were studied by DSC using a Perkin-Elmer Diamond DSC power compensation instrument. The scanning rate was 10 °C min−1 from −50 to 250 °C where approximately 5 mg of each sample was measured and analyzed. Fourier transform infrared spectroscopy (FTIR) spectra were collected with a Perkin Elmer FTIR spectrometer. The polymer and the composites were analyzed as films. The FTIR spectra for polymer and composite films were collected as absorbance spectra using 256 scans at 2 cm−1 resolution. X-ray diffraction (XRD) patterns were recorded on a Siemens XD-500 diffractometer using Cu-Kα radiation. The patterns were recorded over a 2θ range of 5–60° in steps of 0.2° with a counting time of 3 s per step. Samples were in the form of films supported on glass substrates. A microfabricated force device based on the tuning fork was used to measure the mechanical properties of the rGO–PVA composites.30–32 We used a function generator (Model DS345 30 MHz Synthesized Function Generator), and a lock-in amplifier (Model SR830 DSP Lock-in Amplifier) in the laboratory to perform our experiments.Results and discussion
Graphite oxide (GO) is a layered material produced by the oxidation of graphite, which contains many oxygen-containing functional groups to help dispersion at the level of individual sheets. Fig. 1a shows a typical tapping-mode AFM image of GO sheets deposited onto a mica substrate. A line scan of the height profile shows an average topographic height of ∼1.1 nm, consistent with the typical height reported for single-layer GO sheets on mica substrate (1.0 nm). Transmission electron microscopy (TEM) was also employed to determine the existence of individual graphene sheets in water. The TEM image shown in Fig. 1b shows that GO was fully exfoliated into individual sheets by ultrasonic treatment.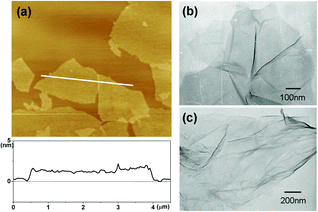 | ||
| Fig. 1 (a) A typical tapping-mode AFM image of graphene sheets deposited on a mica substrate. (b,c) TEM images of an individual GO sheet and rGO sheet. | ||
GO sheets are heavily oxygenated to generate hydroxyl and epoxide functional groups on their basal planes, as well as carbonyl and carboxyl groups located at the sheet edges.11 The FTIR spectrum of GO (Figure S1 in ESI†) shows strong O–H stretch features which appear at 3400 cm−1 as a broad and intense signal. Also, the other characteristic features in the FTIR spectrum of GO are the adsorption bands corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbonyl stretching at 1733 cm−1, the O–H deformation vibration at 1412 cm−1, the C–OH stretching at 1222 cm−1, and the C–O stretching at 1043 cm−1, which confirm the existence of –COOH and –OH groups. The presence of these functional groups makes GO sheets strongly hydrophilic, which allows GO to readily swell and disperse in water (Fig. 2a, left). In this study, the PVA, possessing many –OH groups, was used to protect the graphite oxide because of the hydrogen-bonding interactions between the PVA and GO. As a result, the introduction of this polymer makes graphite oxide not only exfoliate in water but also readily form stable dispersions in polar aprotic solvents (such as DMF) (Fig. 2a, middle).
O carbonyl stretching at 1733 cm−1, the O–H deformation vibration at 1412 cm−1, the C–OH stretching at 1222 cm−1, and the C–O stretching at 1043 cm−1, which confirm the existence of –COOH and –OH groups. The presence of these functional groups makes GO sheets strongly hydrophilic, which allows GO to readily swell and disperse in water (Fig. 2a, left). In this study, the PVA, possessing many –OH groups, was used to protect the graphite oxide because of the hydrogen-bonding interactions between the PVA and GO. As a result, the introduction of this polymer makes graphite oxide not only exfoliate in water but also readily form stable dispersions in polar aprotic solvents (such as DMF) (Fig. 2a, middle).
 | ||
| Fig. 2 (a) An aqueous dispersion of GO sheets (left), a PVA-treated GO dispersion in DMF (middle) and a PVA-treated rGO dispersion in DMF (right). (b) Suspensions of composite samples with different volume fractions of rGO and dissolved in water. | ||
Scheme 1 shows the preparation of rGO–PVA composite hydrogels and wires. In all synthetic routes, keeping the graphene sheets individually separated is the most important and challenging part. Chemical functionalization or the use of dispersants is considered as general method to prevent agglomeration.20 Our way to improve the dispersion of a polymer blend is to incorporate specific chemical interactions between the components. The GO incorporates various oxygen-containing defect sites on the surface and ends, which can interact with the hydroxyl groups of the polymer via hydrogen bonding. We have shown that a true “molecular composite” can be formed by the manipulation of intermolecular hydrogen bonding between the components of polymer and GO, which correlates to the accessibility of the functional groups to participate in intermolecular interactions of polymer composition. Formed by reduction of the PVA-treated GO, we got the rGO-based composites, which showed significant improvements in dispersibility and applicability. Chemically converted graphene sheets obtained from polymer-treated GO can readily form a stable dispersion of individual sheets in organic solvents (Fig. 2a, right), making them more compatible with other materials, facilitating the preparation of composites. Moreover, the mixture of the polymer molecules and rGO sheets not only leads to physical separation of the resultant graphene sheets, but also makes it possible to directly form stable graphene dispersions (Fig. 2b), which could be still easily dispersed with increasing the concentration of the graphene in the composites.
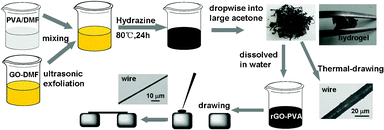 | ||
| Scheme 1 Preparation of rGO–PVA composite hydrogels and wires. The composite wires were made by thermal-drawing or by directly drawing from the rGO–PVA dispersion. | ||
Given that carboxylic acid groups are unlikely to be reduced absolutely by hydrazine under the given reaction conditions, these groups should therefore remain in the reduced product as confirmed by our FTIR analysis (Figure S2 in ESI†). We have obtained elemental analysis data for all the samples (GO, rGO, GO/PVA and rGO–PVA), and these are listed in Table S2 in ESI.† Due to the limited efficiency of the reduction process, the obtained graphene sheets still contained residual oxygenated functional groups of the starting material to protect the graphene dispersion by hydrogen-bonding interactions between the polymer and sheets. It was suggested that GO sheets must be broken into individual sheets in order to achieve effective reinforcement of the polymer matrix. Additionally, it is known from the polymer composite that a strong interaction between filler and matrix is required to transfer stress across the interface. Upon treatment with PVA, the CO stretching vibration at 1733 cm−1 in graphene becomes shifted to lower wavenumbers by the appearance of a stronger absorption at 1703 cm−1 that can be attributed to the carbonyl stretching vibration of the strong interaction between the components.
Polymer composite research pays much attention to enhancing the properties of polymers using molecular or nanoscale reinforcements. The 2D material graphene (Fig. 1c), as a superior nanoscale reinforcement, is expected to have unique mechanical properties due to the ideal graphitic network of sp2 bonds and significantly ordered form.21,22 The properties of a composite are also intimately linked to the aspect ratio and surface-to-volume ratio of the filler. The potential properties of our rGO-based composites thus appear promising owing to the extremely high aspect ratios of the sheets. The dispersion state of the rGO in PVA matrix was detected by SEM images (Fig. 3a,b), in which the graphene maintains the layer structure. The rGO sheets in the composites are crumpled and wrinkled, and at times folded.
 | ||
| Fig. 3 (a) Low and (b) high magnification SEM images obtained from a fracture surface of composite samples of 1.14 vol % rGO in PVA. | ||
Raman spectroscopy has been used to study graphene and graphene-based composites, as both of them possess unique features observed in Raman spectra, where they have a prominent couple of Raman-active bands visible in the spectra (Fig. 4), the D-band observed at ∼1330 cm−1, as well as the G-band appearing at around ∼1590 cm−1.23–25 These bands can be used to detect the presence of graphene in the composites throughout the image on the length scale of the Raman beam size. Both D-band and G-band peaks shifts in the composites relative to those of the graphene can be analyzed to provide an indication of interaction between the graphene and polymer from the Fig. 4 and Table S1 (see ESI†).26 A clear trend in the spectral position of the D-band and G-band before and after the modification with polymer can be noticed. The rGO–PVA composite shows a negative shift in the D peak and a positive shift in the G peak. In the spectra of pristine samples of rGO, the D-band peaks at 1336 cm−1, while in the presence of polymer, this band is seen to shift to lower wavenumbers. The most evident shift is seen in samples of rGO, where the D-band down-shifts by 7 cm−1 from 1336 to 1329 cm−1 and the G-band up-shifts by 8 cm−1 from 1590 to 1598 cm−1 (Fig. 4a). The observed down-shift in the D band and up-shift in the G band could be due to the interaction between the graphene and polymer. Similar results were seen in the cases of the GO–PVA composites (Fig. 4b). Complementarily, the variation of the D-band and G-band intensity in different regions of the composites thus provides quantitative information regarding the dispersion in the sample.
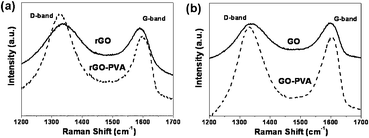 | ||
| Fig. 4 The Raman spectra of rGO and the rGO–PVA composite (a) as well as the Raman spectra of GO and the GO–PVA composite (b). All of the spectra correspond to an exciting laser wavelength of 633 nm. | ||
The association between PVA and rGO sheets has been reported to be quite robust, resulting in the appearance of the shift in the Raman graphene D and G band peak positions due to specific interactions.26 This confirms that there is excellent load transfer between PVA matrix and the rGO. The observed changes in the Raman spectra can be explained by considering wrapping of the polymers around the graphene sheets. The strong attachment of the polymer to the graphene results in the observed down-shift of the D-band and up-shift of the G-band due to increase in the elastic constant of the harmonic oscillator of the polymer-coated the graphene.26 The hydrophobic and van der Waals attraction forces between the polymer and graphene increase the energy necessary for vibrations to occur, which is reflected in the higher frequency of the Raman peaks.27 One should also point out that the spectral shift of the peaks is fairly uniform for the graphene. These two facts are indicative of the homogeneity of the polymer around individual graphene sheets.
Furthermore, the thermal properties of the rGO–PVA composites were studied by DSC, as shown in Fig. 5. The glass transition temperature (Tg) and melt peak (Tm) of the PVA are clearly observed at ∼80 °C and at ∼200 °C. A specific example shown in Fig. 5a is the Tm change from the pure polymer from 192.1 to 218.5 °C for the rGO–PVA composites at 1.8 vol % of graphene, as well as a shift in Tm of 84.6 to 119.2 °C. With increasing volume fraction of graphene, shifts toward higher temperatures of the Tg and Tm are observed, suggesting the degree of polymer crystallization of the rGO–PVA composites is becoming better, as shown in Fig. 5b.28 Similar results were seen in the cases of GO–PVA composites. It should be pointed out that Tm of the graphene composite is much larger than GO composite. These results support our hypothesis of the strong interaction between polymer matrix and nanoscale filler. All composites show DSC curves; however, the characteristic temperatures vary with the amount of graphene in the composition, which correlates with the evidence that indicates that this system also incorporates the most interactions between graphene and polymer matrix. These results are in agreement with previous Raman studies that the specific interaction between polymer matrix and nanoscale filler enables the efficient dispersion of the nanofillers.
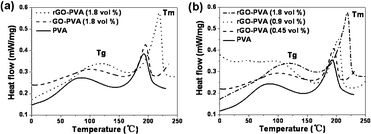 | ||
| Fig. 5 (a) DSC curves for PVA, GO–PVA composite and rGO–PVA composite. (b) DSC curves for a range of rGO–PVA composites with different volume fractions of rGO. Note that the glass transition temperature (Tg) and melt peak (Tm) increase with the graphene content. | ||
We further used XRD to determine whether the rGO sheets were individually dispersed in the rGO-based composites. As shown in Fig. 6, the diffraction peak of GO was found at about 2θ = 10.8°, however, after the GO was dispersed in PVA and was simultaneously reduced, the diffraction peak of rGO–PVA appeared at about 2θ = 19.7°, which only shows the diffraction peak of PVA. These results demonstrate that the GO sheets were individually dispersed in the PVA matrix and maintained the dispersion after reduction, and also provided additional support for successful individually separated graphene without multilayer graphene stacks in the composites.
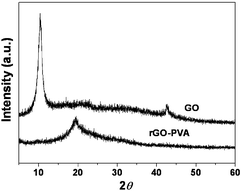 | ||
| Fig. 6 XRD patterns of the GO and rGO–PVA composite. | ||
Here, we used a simple and rapid approach to measure the mechanical properties of the rGO–PVA composite wires (Scheme 1). A drop of PVA and rGO–PVA dispersions was placed on top of one prong which was then quickly drawn into a wire and attached onto the second prong with a sharp metal tip (Figure S3 in ESI†). After evaporation of the solvent, composite wires with graphene embedded in the PVA matrix were formed. Tensile tests were conducted at room temperature using a microfabricated tuning fork (MTF) device.29 The MTF device is made of quartz crystal with two symmetric prongs. The dimensions of each prong are 4 × 0.6 × 0.35 mm, and the separation between the two prongs is ∼200 μm. The unique properties of quartz, optimized crystal cutting angle and symmetric prongs make MTF an extremely sensitive and highly stable force sensor. For example the force sensitivity is ∼1 pN Hz−0.5 and the temperature stability is better than 0.1 ppm °C−1 near room temperature, which is ideal for monitoring subtle changes in the mechanical properties of the composite wire.30
In order to obtain the mechanical properties of the composite wires, we measured the oscillation amplitude of the MTF as a function of frequency (Fig. 7a). From the well defined resonance peaks, we determined the resonance frequency before and after cutting the polymer wire. The resonance frequency (f) with the PVA-composite wire in place was several hundreds of Hz higher than the frequency (f0) with the wire being cut. The difference in the resonance frequencies is related to Young's modulus (E) of the polymer wire and given by
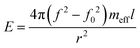 | (1) |
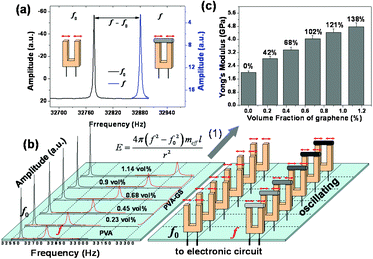 | ||
| Fig. 7 (a) Oscillation amplitude vs. frequency plots showing well defined resonance peaks of a MTF with a micrometer-sized rGO–PVA composite wire (right, f), and after cutting (left, f0). The resonance frequency (f) with the composite wire and the frequency (f0) with the wire being cut were measured under the same conditions. (b) Oscillation amplitude vs. frequency plots showing well defined resonance peaks of the MTF with the composite wire with different volume fractions of graphene, and after cutting. (c), The Young's modulus of the composites with different volume fractions of graphene, the volume fractions of graphene (%) being 0.23, 0.45, 0.68, 0.9, 1.14. Note that the Young's modulus of the composite wire was determined using relation (1). | ||
The typical resonance curves for PVA wires and rGO–PVA composite wires with 0.23, 0.45, 0.68, 0.90, and 1.14 vol % rGO volume ratios given in Fig. 7b and their mechanical properties are shown in Fig. 7c. Substantial increases in mechanical properties are observed as the graphene concentration is increased. Compared to the PVA, as the graphene content is increased to 0.68 vol %, the Young's modulus of rGO–PVA composite increases by ca. 200% compared to the pure polymer from 2 to 4.04 GPa. These results show that the rGO-based composites enable both efficient dispersion and excellent interfacial stress transfer. The significant improvement of the rGO-based composites is probably due to the specific interactions between the components, such as the force of the hydrogen bonding. Furthermore, graphene sheets have high surface-to-volume ratios owing to the accessibility of the atomically thin 2D surface to polymer molecules, which makes graphene sheets potentially more favorable for altering all matrix mechanical properties. We therefore expect that our method for the preparation of individual graphene sheets into polymer matrices by specific interactions will lead to the further development of a broad new class of polymer materials with enhanced properties.34
Conclusions
In summary, our studies have clearly demonstrated that PVA modification is a powerful tool for improving graphene dispersion and enhancing load transfer in their composites. The graphene sheets, which can be effectively functionalized by in situ chemical reduction of the PVA-treated GO, can readily form stable individual sheet dispersions in organic solvents and water and become more compatible with polymer to facilitate the preparation of composites. By directly drawing these dispersions, rGO–PVA composites wires were successfully fabricated with improved dispersion and load transfer in their composites. The mechanical properties of the composite wires were investigated using a MTF device. With a small addition (0.68 vol %) of graphene, the Young's modulus of the PVA wires were reinforced by ca. 200% and their thermal stabilities were increased due to the improved crystallinity of the composites. Raman spectroscopy provided strong evidence of an interaction occurring between the PVA and graphene. We anticipate that this work will highlight a promising direction for the future research and applications of graphene materials.Acknowledgements
This work was financially supported by National Basic Research Program of China (No. 2011CB935704), the National Natural Science Foundation of China (No. 11079002), Research Fund of Ministry of Education of China (No. 20110002130007). We also acknowledge Dr E. Forzani and Dr N. J. Tao (ASU) for help with the tuning fork.References
- D. Li and R. B. Kaner, Science, 2008, 320, 1170 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS.
- J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth and S. Roth, Nature, 2007, 446, 60 CrossRef CAS.
- X. L. Li, X. R. Wang, L. Zhang, S. W. Lee and H. J. Dai, Science, 2008, 319, 1229 CrossRef CAS.
- Y. Zhang, Y. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201 CrossRef CAS.
- S. Gilje, S. Han, M. Wang, K. L. Wang and R. B. Kaner, Nano Lett., 2007, 7, 3394 CrossRef CAS.
- W. Chen, S. Chen, D. C. Qi, X. Y. Gao and A. T. S. Wee, J. Am. Chem. Soc., 2007, 129, 10418 CrossRef CAS.
- D. A. Areshkin and C. T. White, Nano Lett., 2007, 7, 3253 CrossRef CAS.
- S. Stankovich, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342 CrossRef CAS.
- J. M. Michael, L. Je-Luen, H. A. Douglas, C. S. Hannes, A. A. Ahmed, L. Jun, H. Margarita, L. M. David, C. Roberto, K. P. Robert and A. A. Ilhan, Chem. Mater., 2007, 19, 4396 CrossRef.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS.
- S. Park, K.-S. Lee, G. Bozoklu, W. Cai, S. T. Nguyen and R. S. Ruoff, ACS Nano, 2008, 2, 572 CrossRef CAS.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457 CrossRef CAS.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463 CrossRef CAS.
- X. Wang, L. J. Zhi and K. Müllen, Nano Lett., 2008, 8, 323 CrossRef CAS.
- S. Watcharotone, D. A. Dikin, S. Stankovich, R. Piner, I. Jung, G. H. B. Dommett, G. Evmenenko, S. E. Wu, S. F. Chen, C. P. Liu, S. T. Nguyen and R. S. Ruoff, Nano Lett., 2007, 7, 1888 CrossRef CAS.
- (a) Y. Xu, H. Bai, G. Lu, C. Li and G. Shi, J. Am. Chem. Soc., 2008, 130, 5856 CrossRef CAS; (b) J. Liang, Y. Huang, L. Zhang, Y. Wang, Y. Ma, T. Guo and Y. Chen, Adv. Funct. Mater., 2009, 19, 2297 CrossRef CAS; (c) J. Shen, B. Yan, T. Li, Y. Long, N. Li and M. Ye, Soft Matter, 2012, 8, 1831 RSC.
- S. Niyogi, E. Bekyarova, M. E. Itkis, J. L. McWilliams, M. A. Hamon and R. C. Haddon, J. Am. Chem. Soc., 2006, 128, 7720 CrossRef CAS.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS.
- C. G. Lee, X. D. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385 CrossRef CAS.
- C. Gomez-Navarro, M. Burghard and K. Kern, Nano Lett., 2008, 8, 2045 CrossRef CAS.
- N. K. Konstantin, O. Bulent, C. S. Hannes, K. P. Robert, A. A. Llhan and C. Roberto, Nano Lett., 2008, 8, 36 CrossRef.
- I. Calizo, A. A. Balandin, W. Bao, F. Miao and C. N. Lau, Nano Lett., 2007, 7, 2645 CrossRef CAS.
- D. Graf, F. Molitor, K. Ensslin, C. Stampfer, A. Jungen, C. Hierold and L. Wirtz, Nano Lett., 2007, 7, 238 CrossRef CAS.
- A. S. Vladimir, K. G. Muhammed, A. Y. Alexander, A. R. Anna, S. Kai, A. M. Arif, P. W. James and A. K. Nicholas, J. Am. Chem. Soc., 2005, 127, 3463 CrossRef.
- R. Asif, D. D. Mark, I. Ilia, F. B. Phillip and B. G. David, Chem. Mater., 2006, 18, 3513 CrossRef.
- M. Cadek, J. N. Coleman, K. P. Ryan, V. Nicolosi, G. Bister, A. Fonseca, J. B. Nagy, K. Szostak, F. Béguin and W. J. Blau, Nano Lett., 2004, 4, 353 CrossRef CAS.
- S. Boussaad and N. J. Tao, Nano Lett., 2003, 3, 1173 CrossRef CAS.
- M. H. Ren, E. S. Forzani and N. J. Tao, Anal. Chem., 2005, 77, 2700 CrossRef CAS.
- S. J. O'Shea and M. E. Weland, Langmuir, 1998, 14, 4186 CrossRef CAS.
- F. Tsow and N. J. Tao, Appl. Phys. Lett., 2007, 90, 174102 CrossRef.
- J. Zhang and S. O'Shea, Sens. Actuators, B, 2003, 94, 65 CrossRef.
- X. F. Zhang, T. Liu, T. V. Sreekumar, S. Kumar, V. C. Moore, R. H. Hauge and R. E. Smalley, Nano Lett., 2003, 3, 1285 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR of GO, PVA and rGO–PVA, Raman peak positions of rGO and rGO–PVA composites, mechanism of the mechanical properties measurement, SEM image of the modified tuning fork with a single polymer wire. See DOI: 10.1039/c2ra20644g |
| This journal is © The Royal Society of Chemistry 2012 |