In situ preparation of silver nanoparticles on biocompatible methacrylated poly(vinyl alcohol) and cellulose based polymeric nanofibers†
Narahari
Mahanta
and
Suresh
Valiyaveettil
*
Department of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543. E-mail: chmsv@nus.edu.sg
First published on 26th September 2012
Abstract
Ultra-fine nanofiber mats of cellulosic polymer and polyvinyl alcohol (PVA) containing silver (Ag) nanoparticles were fabricated using the electrospinning technique. Polymers with hydroxyl groups on the backbone such as PVA, PVA/dextran and PVA/methyl cellulose were chosen for this study. All polymers showed the ability to reduce silver nitrate (AgNO3) to Ag-nanoparticles. To make crosslinkable networks of the water soluble polymers, the photo-crosslinkable glycidyl methacrylate moiety was attached to the hydroxyl groups of the designed macromers. AgNO3 and cytocompatible initiator (IRGACURE 2959) were added to the methacrylated polymeric solution mixture before electrospinning. The fiber morphology of the resultant nanofiber was characterised using scanning electron microscopy (SEM) and an average diameter in the range of 300 to 500 nm was observed. The resultant nanofibers containing Ag-ions and initiators were crosslinked under photoirradiation at a wavelength of 365 nm for 6 h and heated at 100 °C for 6 h for complete reduction of Ag-ions into Ag-nanoparticles. Transmission electron microscopy (TEM) analysis revealed that the Ag-nanoparticles with an average diameter of 10 to 20 nm were uniformly distributed throughout the nanofibers and its presence was confirmed using X-ray diffraction analysis. The effect of Ag-nanoparticles on the microcrystalline structure of the polymer was studied by thermal and mechanical properties analysis. The biocompatibility of the methacrylated macromers was investigated using human lung fibroblasts (IMR-90) and no cytotoxic effects were found after 4 days of incubation. The antimicrobial activity of the PVA nanofiber containing Ag-nanoparticles was tested against Escherichia coli. The nanofiber containing Ag-nanoparticles may have potential applications in biomedical devices which will be explored in the near future.
Introduction
Silver-polymer nanostructured materials have immense implications in biology,1 catalysis2 and antimicrobial applications.3–6 Several fabrication techniques have been used for the fabrication of non-agglomerated nanoparticles with a polymer as the template.7–12 Silver (Ag) nanoparticle loaded fabrics have great potential in the fields of antiseptic dressings, bandages and for biomedical applications. Owing to that, hydrophilic polymeric hydrogels with Ag-nanoparticles were prepared and applied for antimicrobial applications.13–16 Nanofibers prepared using electrospinning have attracted attention because of their highly porous polymeric network with superior mechanical properties and high surface area to volume ratio.17,18 As a result, such nanofibers were used to fabricate polymer nanofiber mats for different applications.19–22A few mechanically stable and biodegradable macromers containing Ag-nanoparticles were used to prepare nanofibers and showed antimicrobial activities against S. aureus and E. coli.23
Recently many researchers fabricated natural polysaccharides and applied them in various biomedical and other advanced applications24–27 because of their widely useful properties such as nontoxicity, biocompatiblity and biodegradablity.28–30 In addition, cellulose based polymers were also used for synthesis and capping agents for metal nanoparticles.4,31,32 Cellulose contains a number of hydroxyl groups with lone pairs of electrons which help to create ion–dipole linkages, hence stabilizing the Ag-nanoparticles without coagulation. Poor mechanical properties33 and easy biodegradability34 of such natural polymers limit their use in many applications. To overcome these issues several researchers have focused on fabrication of hybrids from mechanically stable polymers, such as polystyrene and polycaprolactone with Ag-nanoparticles.3,35,36 Furthermore, poly(methyl methacrylate) nanofiber containing Ag-nanoparticles was synthesized by radical mediated dispersion polymerization and applied as an antibacterial agent.15 Ag-nanoparticles incorporated in the polymer nanofiber showed enhanced antibacterial efficacy compared to that of silver sulfadiazine and silver nitrate at the same silver concentration.
In the present study, hydrophilic biocompatible polymers with functional groups that complex with silver and sufficient mechanical integrity were chosen as a polymer matrix. Photocrosslinkable PVA and polysaccharide based polymers (dextran or methyl cellulose) were used to fabricate a few nanofibrous mats with different compositions via the electrospinning technique. Polysaccharides with hydroxyl groups can efficiently coordinate nanoparticles and mixing with atactic PVA provides good mechanical integrity. The effect of Ag-nanoparticles on the microcrystalline structure of the polymer matrix was investigated by thermal and mechanical analysis. The biocompatibility of the designed methacrylated nanofiber was tested using human lung fibroblast IMR-90 and the results are discussed in detail. Furthermore, the antimicrobial activity of the PVA–Ag NF was tested against Escherichia coli by a zone inhibition assay.
Experimental
Materials and methods
Polyvinyl alcohol (PVA) (Mw ≈ 146![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–186000), dextran (DEX, Mw ≈ 35000–45000), silver nitrate (99.99%), triethylchlorosilane, glycidyl methacrylate (GMA) and 4-dimethylaminopyridine (DMAP) were purchased from Sigma Aldrich, Singapore. Methyl cellulose (MC) was purchased from BDH chemicals ltd. England. All chemicals were used without further purification. Prior to experiment, all glassware was thoroughly washed and dried.
000–186000), dextran (DEX, Mw ≈ 35000–45000), silver nitrate (99.99%), triethylchlorosilane, glycidyl methacrylate (GMA) and 4-dimethylaminopyridine (DMAP) were purchased from Sigma Aldrich, Singapore. Methyl cellulose (MC) was purchased from BDH chemicals ltd. England. All chemicals were used without further purification. Prior to experiment, all glassware was thoroughly washed and dried.
Aqueous PVA solution (10 wt%) was prepared by heating at 80 °C with continuous stirring. Similarly MC (5 wt%) and DEX (5 wt%) solutions were prepared by dissolving in ultrapure water at room temperature with constant stirring for 12 h. Solutions of PVA/MC (10:5) and PVA/DEX (10:5) were prepared by mixing the prepared individual polymer solutions and were kept under constant stirring for 6 h at ambient conditions. AgNO3 was dissolved in deionised water in the dark to avoid the chance of reduction under light. An appropriate amount of AgNO3 (5 wt%) was added to the polymer solution and stirred for 5 min for proper homogeneity. Fig. 1 shows the schematic representation of the procedure followed for the electrospinning of Ag/polymer nanofibers.
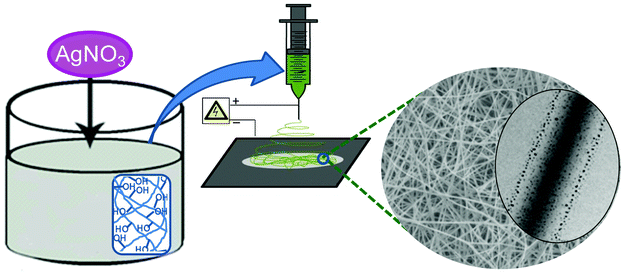 | ||
| Fig. 1 Schematic diagram for the preparation of polymer nanofibers containing silver nanoparticles by the electrospinning technique. | ||
To prepare the hydrophobic surface, glass cover slips were immersed in triethyl chlorosilane and kept for 5 min. The dipping process was performed twice, then the surface was rinsed with water and finally dried under RT before using it for nanofiber collection.
Preparation of methacrylated poly(vinyl alcohol)/dextran/methyl cellulose
Methacrylated PVA was prepared according to the procedure proposed by Hennink et al.37,38 PVA (2 g) was dissolved in dimethyl sulfoxide (DMSO) (50 ml) and DMAP (1 g) was added to it. GMA was added in the proper molar ratio with respect to the number of hydroxyl groups (based on per monomer unit) and the reaction mixture was stirred for 2 days at RT. The reaction was carried out under a nitrogen atmosphere. A similar procedure was used for the preparation of methacrylated methyl cellulose and methacrylated dextran. After the reaction has finished, the substituted products were dialysed against water, reprecipitated with acetone and freeze dried. The final products were stored at −20 °C to avoid self-crosslinking. The methacrylated derivatives obtained from all designed macromers were characterized using nuclear magnetic resonance (NMR) spectroscopy.Test for the reduction of silver salt
The color change in the reaction medium was used to monitor the extent of reduction of AgNO3 by different polymers (and their mixtures) such as PVA (10 wt%), DEX (5 wt%), MC (5 wt%), PVA (10 wt%) + DEX (5 wt%) and PVA (10 wt%) + MC (5 wt%). AgNO3 solution (5 wt%) was added to each polymer mixture and mixed gently. Camera images of color change experiments were taken at different time intervals such as 0 h, 6 h, 20 h and 48 h.Fabrication of polymer nanofibers containing Ag-nanoparticles
For our study, aqueous AgNO3 was added to the solution of methacrylated macromers and stirred for 5 to 10 min before electrospinning. AgNO3 (5 wt%) corresponding to the total dry weight of polymer was added to all polymer solution. The electrospinning setup consists of a hypodermic syringe with a needle (internal diameter of 1.2 mm) connected to a programmable syringe pump (Optrobio Technologies Pte Ltd) was used to control the syringe. The positive electrode of the high voltage power supply was connected to the syringe needle and negative end was connected to the grounded copper plate covered with aluminum foil which served as the collector. The solutions were electrospun at a positive voltage supply of 15 to 18 kV. The distance between the needle tip and collecting plate was kept at 10 cm and a constant flow rate was maintained at 4 μl min−1. The nonwoven electrospun nanofibers were collected on the surface modified glass cover slips placed on the electrically grounded aluminum foil. The whole process was carried out at ambient conditions. During the electrospinning process, the fibers were brownish in colour, indicating reduction of Ag-ion to Ag-nanoparticles. The resultant nanofibers were kept under UV irradiation at 365 nm for crosslinking for 6 h. The samples were further heated at 100 °C for 6 h inside the oven for complete reduction of Ag-ions.Cytotoxicity analysis of the designed methacrylated macromers
Electrospun nanofiber sheets derived from methacrylated macromers (PVA MA, PVA/DEX MA and PVA/MC MA) were collected on round glass cover slips (diameter 13 mm) and then crosslinked as discussed in above. Then the fiber samples were placed on 24 well culture plates and conditioned in sterile phosphate buffered saline (PBS) before seeding with cells. IMR-90 (passage 18 ± 3) was purchased from Coriell Cell Repositories, USA and maintained in modified eagles medium with glutamine supplemented with 15% fetal bovine serum (FBS, Standard Quality, EU-approved origin PAA Laboratories), 1% each of penicillin, streptomycin, non-essential amino acids, vitamins, and 2% essential amino acids (Gibco, Invitrogen). Cells were subcultured at 80–90% confluence and maintained at 37 °C in a humidified incubator with 5% CO2 gas.The viability of cells on the respective nanofiber mesh was measured using the CellTiter-Glo luminescent cell viability assay (Promega) following the manufacturer's instructions. For the viability assay, ∼5000 cells were placed along with the nanofibers in a 24-well plate (Griener Bio-one). The cells were incubated for 4 days in 1 ml medium per well at 37 °C in a humidified incubator with 5% CO2. At respective timings, the medium in each well was removed and replaced with 200 μl fresh medium. CellTiter-Glo (200 μl) reagent, pre-warmed to room temperature was then added and mixed properly. After 10 mins, the mixture was transferred to flat-bottom 96-well plates (Corning, Costar) and luminescence readings were recorded using a Tecan Infinite F200 micro-plate reader.
Antimicrobial tests
The PVA nanofibers with and without Ag nanoparticles were prepared on the round shaped glass cover slip and were sterilized by UV irradiation prior to investigation for antimicrobial activity. The antimicrobial activity of the designed nanofibers was tested against Escherichia coli by the zone inhibition assay. Briefly, overnight grown bacterial cells in Luria-Bertani (LB) medium were sub-cultured to get exponential phase culture. The density of the cells was then adjusted to 106 CFU/ml. A cotton swab was dipped in and was streaked onto a LB agar plate. The pre-sterilized PV NF and the PVAg NF were placed carefully on the bacterial cultured plate and were incubated at 37 °C for 16 h and the inhibitory zone was examined under our working conditions.Characterization
NMR spectra of the methacrylated derivatives were recorded using a Bruker 400 MHz instrument with tetramethylsilane (TMS) as an internal standard and d-DMSO as the solvent. The resultant fiber morphology of the nanofiber was observed using field emission scanning electron microscopy (FESEM, JEOL JSM-6701F). The electrospun nanofibers were collected on glass cover slips and were mounted on copper stubs with a double sided conducting carbon tape coated with platinum. Similarly, the electrospun samples were deposited on carbon coated copper grids and used for transmission electron microscopy (TEM) study using a JEOL JEM 2010 with an accelerating voltage of 200 kV. X-Ray diffraction (XRD) patterns were taken with a Bruker-AXS D8 DISCOVER with a GADDS powder X-ray diffractometer in a 2θ scan configuration in the range of 2° to 70°. Thermal properties of the composite nanofibers were studied using a thermogravimetric analyser (TGA) (TA instrument 2960) from room temperature to 900 °C and differential scanning calorimetry (DSC) (TA instrument 2920) at a heating rate of 10 °C min−1 in an inert N2 atmosphere. For the DSC experiment aluminum sample pans were used as sample holders. Static mechanical characterization was carried out by a dynamic mechanical analyser (DMA) instrument (TA instrument, Q800). Rectangular shaped nanofiber strips of 3 mm × 10 mm were used for the tensile testing. The nanofiber was clamped on the tension clamp and the DMA controlled force mode was used. The preload force of 0.001 N was applied and the temperature of the chamber was maintained at 20 °C during experimentation. The force was ramped at a rate of 0.01 N min−1 and the mechanical properties were recorded.Results and discussion
Test for the reduction of silver salt by the designed polymers
The extent of the reduction of silver nitrate was observed by a colour changing experiment for different polymers and mixtures (Fig. 2). The extent of the reduction of Ag-ions to Ag-nanoparticles with the designed polymer solution can be observed directly and depends on the functionality present on the polymer backbone. It is conceivable that the hydroxyl groups of PVA, DEX and methyl cellulose are the key functional groups responsible for the reduction of Ag-ions to Ag-nanoparticles. | ||
| Fig. 2 Test for the reduction of AgNO3 into Ag-nanoparticles, with different polymer solutions (PVA 10 wt% (A), DEX 10 wt% (B), MC 5 wt% (C), PVA 10 wt% + DEX 5 wt% (D), PVA 10 wt% + MC 5 wt% (E)) at the time intervals of 0 h, 6 h, 20 h and 48 h. | ||
The hydroxyl groups having lone pairs of electrons, coordinate with Ag-ions and act as a reducing agent as well as a stabilizer for the Ag-nanoparticles.39 A brown coloration was observed after a certain period of time indicating that the reduction of the Ag-ions and subsequent formation of Ag-nanoparticles increased with time. It was observed that the rate of particle formation was maximum for the case of methyl cellulose as compared to PVA and dextran with the same amount of AgNO3 loading. Furthermore, the color for PVA–AgNO3 remained fairly clear throughout the experiment. The low reduction of Ag+ ions in the PVA/DEX–AgNO3 mixture was due to the low availability of hydroxyl groups as a result of intermolecular hydrogen bonds between DEX and PVA molecules.
The presence of methoxy groups on MC results in weak inter-polymer chain interactions in aqueous medium. In addition, methoxy groups in MC are weak hydrogen bond acceptors, which in turn increases the availability of free –OH groups on the PVA backbone for facilitating the reduction of Ag cations. Colour changes during the reduction process provide information on the extent of the formation of Ag-nanoparticles on the fiber surface.
Designing the methacrylated macromers
As our designed macromers are water soluble, it is important to make them water insoluble for real applications. A few crosslinking experiments were used to reduce the water solubility for various hydrophilic polymers.40,41 But many of the crosslinking agents are highly toxic in nature.42 Owing to that, an alternate crosslinking method was developed by attaching methacrylated groups to the hydroxyl groups of the macromer. For this, the methacrylate group was introduced to PVA, DEX and MC by reacting with GMA in presence of DMAP in DMSO as a solvent.43,44 The pendent methacrylated groups of the macromers were crosslinked via photopolymerization. For crosslinking, a cytocompatible initiator, IRGACURE 2959 (1 wt%) was mixed with the solution prior to electrospinning. The NMR spectrum of the methacrylated-macromer showed peaks of vinyl protons at δ 5.6 and δ 5.9 ppm, the methyl group showed a peak at δ 1.8 ppm and the anomeric proton appeared at δ 5.2 ppm (Fig. S1 ESI†).Nanofibers of the methacrylated macromers by the electrospinning technique
Electrospinning was used to prepare highly porous nonwoven nanofibrous mats. The mixture of methacrylated macromers, AgNO3 and initiator was mixed in a proper ratio and electrospun into the nanofibrous structure. The preparation of nanofibers by the electrospinning process entails the utilization of a high voltage to create an electrically charged jet of polymer solution. An oppositely charged power supply was connected to the needle and grounded collector plate. After a critical voltage, the electrostatic force overcomes the surface tension of the polymeric solution and the charged jet is ejected from the tip of the needle which undergoes a process of elongation and evaporation leaving behind a nonwoven nanofiber mat. During the electrospinning process, in presence of high voltage, Ag ions were converted to Ag-nanoparticles inside the fiber matrix with an observable color change.To get the optimised nanofiber without any bead formation, different polymer concentrations and blend compositions were used for electrospinning. It is well known that the properties of the polymer solutions such as viscosity, surface tension, electrical conductivity, polymer concentrations and other parameters strongly influence the formation of nanofibers during the electrospinning process. The changes in the viscosity, or conductivity did not lead to the formation of MC nanofibers. PVA (10 wt%) was added to a solution of MC (5 wt%) in the volume ratio of 1:1 for getting smooth nanofibers. Similarly, PVA (10 wt%) was added to DEX (5 wt%) and electrospun to get smooth nanofibers without any bead formation. The morphologies of the PVA–Ag, PVA/DEX–Ag and PVA/MC–Ag fibers were observed under similar conditions using SEM (Fig. 3). All nanofibers showed diameters ranging from 300 to 500 nm. For the case of PVA–Ag, fibers with smooth morphologies were obtained but PVA/MC–Ag and PVA/DEX–Ag fibres showed more interconnections and were not as smooth as the PVA–Ag nanofibers.
 | ||
| Fig. 3 Scanning electron microscopy images of PVA–Ag nanofiber (A), PVA/DEX–Ag nanofiber (B) and PVA/MC–Ag nanofiber (C). | ||
The presence of methacrylate groups on the macromer and initiator in the nanofibers helps to crosslink the polymer chains in the solid state. The nanofibers were irradiated under UV (365 nm) for 6 h, and the reduction of Ag-ions was completed by heating at 100 °C for 6 h. During these stages traces of water were also removed from the fiber matrix. The formation of Ag-nanoparticles was observed under TEM (Fig. 4). It was found that the distribution of spherical Ag-nanoparticles with an average diameter of 10 to 20 nm was uniform throughout the nanofiber matrix.
 | ||
| Fig. 4 Transmission electron microscopy images of PVA–Ag nanofiber (A), PVA/DEX–Ag nanofiber (B) and PVA/MC–Ag nanofiber (C). In the TEM image (A) an inset image shows the high magnification image of nanoparticles on the PVA-nanofiber. | ||
Thermal analysis of the designed macromers with Ag-nanoparticles
The purpose of crosslinking the macromer is to make the entire fiber insoluble in water and to enhance mechanical integrity. Thermal characteristics during dynamic processing conditions provide information on stability, shrinkage, expansion and storage. The intrinsic chain flexibility and temperature required to induce segmental motion is determined by the nature and amount of crosslinks. Simultaneously the introduction of Ag-nanoparticles in the polymer matrix also effects the segmental motion of the polymer backbone. To gain a better understanding on the effect of crosslinking and nanoparticles towards the molecular mobility of the designed macromer, thermal properties of the composite nanofiber were analysed using differential scanning calorimetry (DSC). DSC traces of PVA NF, PVA-DEX NF and PVA/MC NF with and without Ag-nanoparticles were shown in Fig. 5. All values are recorded for the second heating cycles. From the figure, it can be seen that the glass transition temperature (Tg) of PVA nanofiber is 71.2 °C and that of PVA/DEX NF showed a Tg at 79.9 °C. This could be due to the hydrogen bond interaction between PVA and the DEX moiety which increases the glass transition temperature. For the case of PVA/MC, the Tg was observed at 72.8 °C, which indicates significant interaction between PVA and MC. A decrease in Tg by the incorporation of Ag-nanoparticles was observed for all polymeric nanofibers.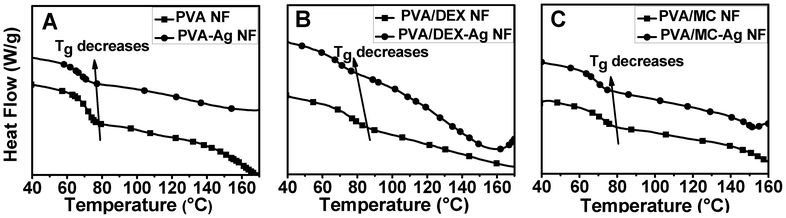 | ||
| Fig. 5 Effect of Ag-nanoparticles on the thermal properties of PVA NF (A), PVA/DEX NF (B), and PVA/MC NF (C). | ||
The incorporation of Ag-nanoparticles led to a decrease in the interaction between polymer chains and an increase in free volume inside the lattice, which led to a decrease in Tg. Similarly, thermal degradation behaviour of the polymer blends mixed with Ag-nanoparticles was carried out by TGA analysis (Fig. S2 ESI†). The first weight loss was due to the loss of adsorbed moisture and volatile materials inside the composite nanofibers. A second decomposition was observed at a range of 250 °C to 370 °C due to the degradation of PVA via the dehydration reaction of polymer chains. The third weight loss between 370 °C and 450 °C corresponds to further degradation of the polymer backbone. In the case of PVA, PVA/DEX and PVA/MC nanofibers containing Ag-nanoparticles, the residual content was maximum even after heating upto 900 °C.
X-Ray diffraction study of Ag-nanoparticle incorporated nanofibers
The formation of Ag-nanoparticles in the nanofiber matrix was further confirmed by X-ray diffraction analysis (Fig. 6). Five peaks were observed at 2θ = 38.4°, 44.2°, 64.5°, 77.6° and 81.6°, which corresponds to the crystal faces of (111), (200), (220), (311) and (222) of silver, consistent with the formation of face-centred-cubic silver crystallite inside the nanofiber matrix.45,46 This peak was compared with the standard powder diffraction card of the Joint Committee on Powder Diffraction Standards (JCPDS), silver file No. 04–0783.47 Since PVA is semicrystalline and both MC and DEX are amorphous in nature, the small intensity diffraction peaks of the polymers are dominated by the presence of sharp peaks from Ag- nanoparticles.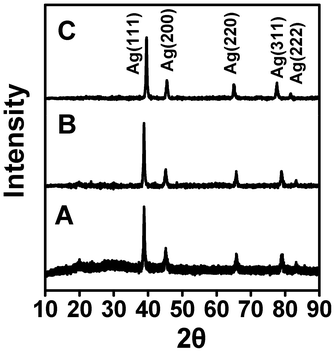 | ||
| Fig. 6 X-Ray diffraction (XRD) pattern of PVA–Ag nanofiber (A), PVA/DEX–Ag nanofiber (B) and PVA/MC–Ag nanofiber (C). | ||
Mechanical properties of the designed macromers with Ag-nanoparticles
The mechanical properties of fibers in the static condition need to be evaluated because of the load bearing capacity plays an important role during real applications. For this, uniaxial stress–strain behavior of the designed nanofibers was investigated using the tensile mode. This measurement provides information on material properties which includes the tensile (Young's) modulus, tensile strength (TS) (ultimate or at break) and also the elongations at break (EB). The data from mechanical studies of PVA NF and Ag-nanoparticle incorporated nanofibers are shown in Table 1.From the data, the modulus, TS and EB decreases with the incorporation of Ag-nanoparticles for all nanofibers. The TS was decreased from 12.6 MPa for PVA NF to 8 MPa for PVA/DEX–Ag nanofibers. Similarly for the case of PVA/MC–Ag nanofibers, it was decreased to 4.6 MPa. Significant differences in modulus, TS and EB between PVA/DEX–Ag NF and PVA/MC–Ag NF were also observed.
The decrease in mechanical properties is due to the effect of nanoparticles on the nanofiber matrix which can be correlated with an increase in volume fraction and the nature of the nanoparticles. During the application of tensile force to the polymer matrix, the nanoparticle polymer interface regions pull apart physically resulting in the formation of many molecular level cracks and voids. For the polymers, the fracture at the interface depends on the forces of attachment of fillers, the packing mechanism and any influence on molecular orientation. As the nanoparticles are in the range of 10–20 nm in size, with weak interactions with the matrix it can be concluded that the Ag-nanoparticle offers no reinforcing action towards the nanofiber matrix which led to a decrease in the mechanical properties.
Cytotoxic analysis on the designed nanofibers and the antimicrobial activity of PVA–Ag nanofibers
For biomaterial applications, a few criteria such as suitable functional groups, physico-mechanical properties, expected durability and suitable interface properties must be considered. Apart from being biocompatible, the material should be free from toxic leachable chemicals during the application.All methacrylated PVA, dextran and MC nanofibers were screened for cytotoxicity analysis and cell viability assay up to 4 days with the commercially available fibroblast cell line (IMR-90). After 4 days, the cell morphology was found to be normal (Fig. 7A) indicating that the fibrous material showed no cytotoxic effect towards the surrounding cells. In a typical procedure, all methacrylated nanofibers (10 mm diameter) were placed over the 24-well culture plate with IMR-90 cells. The plates were incubated at 37 °C and the cell viability was monitored for a few days. The cell viability data revealed no enhancement or reduction in cell growth in presence of methacrylated nanofibers in comparison to the control after 4 days of incubation (Fig. 7B). These results suggest that the methacrylated nanofibers showed no cytotoxic effect towards human cells.
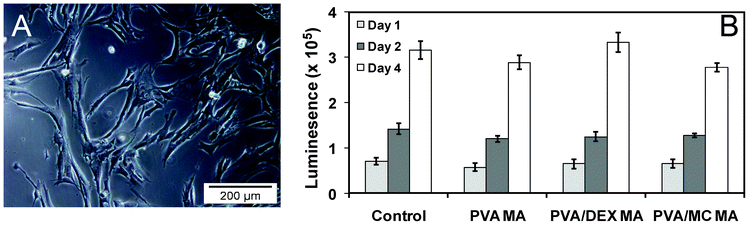 | ||
| Fig. 7 Optical micrograph of IMR-90 cells around nanofiber after 4 days of coculture (A). Cell viability test with IMR-90 on the designed nanofiber up to 4 days of analysis (B). The data represented the mean and standard deviations of the four samples. The control experiment was normalised as the same cell line was used with different time scales. | ||
The antimicrobial activity of PVA and PVA–Ag nanofibers was examined against E. coli (Fig. S3 ESI†). From the data, it was observed that the pure PVA nanofiber did not show any bacterial growth inhibition. However the PVA–Ag nanofiber showed an inhibitory zone around the nanofiber after 16 h of incubation at 37 °C. This preliminary result indicates antimicrobial activity towards the E. coli, which is in good agreement with previously reported literature.23,47,48
Conclusion
The present investigation is focused on the preparation of ultrafine nanofibrous membranes from water soluble polymers incorporated with Ag-nanoparticles prepared using the electrospinning technique. To make the water soluble polymer insoluble in aqueous environment, photocrosslinkable methacrylate groups were incorporated on the polymer backbone. These nanofibers were photocrosslinked under UV irradiation and annealed at 100 °C for the reduction of Ag-ions to Ag-nanoparticles. Characterization of the PVA–Ag, PVA/DEX–Ag, and PVA/MC–Ag nanofibers was carried out using various physico-mechanical analyses. The average diameter of the nanofibers ranged between 300 to 500 nm. TEM revealed that Ag- nanoparticles with an average diameter of 10 to 20 nm are uniformly distributed throughout the nanofibers. Furthermore, powder XRD confirms the presence of Ag-nanoparticles in the nanofiber matrix. The effect of Ag-nanoparticles on modifying the thermal properties of the hybrid nanofibers was evaluated using DSC and stress–strain measurements. The effectiveness of the silver nitrate reduction was improved with the addition of dextran and methyl cellulose separately to the PVA solution. The cytocompatibility nature and antimicrobial activity of PVA–Ag nanofibers suggest that our designed nanofibers may be applicable in biomedical research.Acknowledgements
NM thanks the National University of Singapore (NUS) for graduate scholarship and help from Teow Yiwei in the cytotoxic evaluation of nanofibers. SV thank the Environment and Water Industry Programme Office (EWI) under the National Research Foundation of Singapore (PUBPP 21100/36/2, NUS WBS no. R-706-002-013-290, R-143-000-458-750, R-143-000-458-731) for the financial support of the work. Technical support from the Department of Chemistry, NUS, is greatly appreciated.References
- J. Chen, F. Saeki, B. J. Wiley, H. Cang, M. J. Cobb, Z. Y. Li, L. Au, H. Zhang, M. B. Kimmey, X. D. Li and Y. Xia, Nano Lett., 2005, 5, 473–477 CrossRef CAS.
- A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757–3778 CrossRef CAS.
- H. Kong and J. Jang, Biomacromolecules, 2008, 9, 2677–2681 CrossRef CAS.
- W. K. Son, J. H. Youk and W. H. Park, Carbohydr. Polym., 2006, 65, 430–434 CrossRef CAS.
- M. M. Stevanovic, S. D. Skapin, I. Bracko, M. Milenkovic, J. Petkovic, M. Filipic and D. P. Uskokovic, Polymer, 2012, 53, 2818–2828 CrossRef CAS.
- C. W. Wang, S. J. Tseng, S. F. Peng, Y. K. Hwu and C. K. Lin, Nanotechnology, 2012, 23 Search PubMed.
- R. Mallampati and S. Valiyaveettil, J. Nanosci. Nanotechnol., 2012, 12, 618–622 CrossRef CAS.
- W. J. Liu, Z. C. Zhang, H. R. Liu, W. D. He, X. W. Ge and M. Z. Wang, Mater. Lett., 2007, 61, 1801–1804 CrossRef CAS.
- J. H. He, T. Kunitake and T. Watanabe, Chem. Commun., 2005, 795–796 RSC.
- P. B. Gupta and M. Bajpai, J. Cotton Sci., 2008, 12, 280–286 CAS.
- M. Yanilmaz, F. Kalaoglu, H. Karakas and A. S. Sarac, J. Appl. Polym. Sci., 2012, 125, 4100–4108 CrossRef CAS.
- Y. H. Zhao, Y. Zhou, X. M. Wu, L. Wang, L. Xu and S. C. Wei, Appl. Surf. Sci., 2012, 258, 8867–8873 CrossRef CAS.
- J. Song, M. L. Chen, V. R. Regina, C. X. Wang, R. L. Meyer, E. Q. Xie, C. Wang, F. Besenbacher and M. D. Dong, Adv. Eng. Mater., 2012, 14, B240–B246 CrossRef.
- Y. M. Mohan, K. Lee, T. Premkumar and K. E. Geckeler, Polymer, 2007, 48, 158–164 CrossRef CAS.
- H. Kong and J. Jang, Langmuir, 2008, 24, 2051–2056 CrossRef CAS.
- J. J. Miao, R. C. Pangule, E. E. Paskaleva, E. E. Hwang, R. S. Kane, R. J. Linhardt and J. S. Dordick, Biomaterials, 2011, 32, 9557–9567 CrossRef CAS.
- N. Mahanta and S. Valiyaveettil, Nanoscale, 2011, 3, 4625–4631 RSC.
- S. Ramakrishna, K. Fujihara, W. E. Teo, T. Yong, Z. W. Ma and R. Ramaseshan, Mater. Today, 2006, 9, 40–50 CrossRef CAS.
- K. D. A. Almeida, A. A. A. de Queiroz, O. Z. Higa, G. A. Abraham and J. S. Roman, J. Biomed. Mater. Res., 2004, 68A, 473–478 CrossRef CAS.
- A. Melaiye, Z. H. Sun, K. Hindi, A. Milsted, D. Ely, D. H. Reneker, C. A. Tessier and W. J. Youngs, J. Am. Chem. Soc., 2005, 127, 2285–2291 CrossRef CAS.
- Z. L. Shi, K. G. Neoh and E. T. Kang, Langmuir, 2004, 20, 6847–6852 CrossRef CAS.
- N. Mahanta, Y. Teow and S. Valiyaveettil, J. Nanosci. Nanotechnol., 2012, 12, 6156–6162 CrossRef CAS.
- X. Y. Xu, Q. B. Yang, Y. Z. Wang, H. J. Yu, X. S. Chen and X. B. Jing, Eur. Polym. J., 2006, 42, 2081–2087 CrossRef CAS.
- N. Mahanta, W. Y. Leong and S. Valiyaveettil, J. Mater. Chem., 2012, 22, 1985–1993 RSC.
- A. Autissier, C. Le Visage, C. Pouzet, F. Chaubet and D. Letourneur, Acta Biomater., 2010, 6, 3640–3648 CrossRef CAS.
- J. Kim, Z. J. Cai, H. S. Lee, G. S. Choi, D. H. Lee and C. Jo, J. Polym. Res., 2011, 18, 739–744 CrossRef CAS.
- R. Barbucci, R. Giardino, M. De Cagna, L. Golini and D. Pasqui, Soft Matter, 2010, 6, 3524–3532 RSC.
- P. N. Galgut, Biomaterials, 1990, 11, 561–564 CrossRef CAS.
- N. Villandier and A. Corma, Chem. Commun., 2010, 46, 4408–4410 RSC.
- H. A. Wege, S. Kim, V. N. Paunov, Q. X. Zhong and O. D. Velev, Langmuir, 2008, 24, 9245–9253 CrossRef CAS.
- N. D. Luong, Y. Lee and J. D. Nam, Eur. Polym. J., 2008, 44, 3116–3121 CrossRef CAS.
- C. Y. Zhu, J. F. Xue and J. H. He, J. Nanosci. Nanotechnol., 2009, 9, 3067–3074 CrossRef CAS.
- J. T. Oliveira and R. L. Reis, J. Tissue Eng. Regener. Med., 2011, 5, 421–436 CrossRef CAS.
- M. I. Santos, S. Fuchs, M. E. Gomes, R. E. Unger, R. L. Reis and C. J. Kirkpatrick, Biomaterials, 2007, 28, 240–248 CrossRef CAS.
- J. P. Chen and Y. Chiang, J. Nanosci. Nanotechnol., 2010, 10, 7560–7564 CrossRef CAS.
- R. Nirmala, K. T. Nam, D. K. Park, B. Woo-il, R. Navamathavan and H. Y. Kim, Surf. Coat. Technol., 2010, 205, 174–181 CrossRef CAS.
- W. N. E. van Dijk-Wolthuis, O. Franssen, H. Talsma, M. J. van Steenbergen, J. J. Kettenes-van den Bosch and W. E. Hennink, Macromolecules, 1995, 28, 6317–6322 CrossRef CAS.
- W. N. E. van Dijk-Wolthuis, J. J. Kettenes-van den Bosch, H. A. van der Kerk-van and W. E. Hennink, Macromolecules, 1997, 30, 3411–3413 CrossRef CAS.
- H. Kong and J. Jang, Chem. Commun., 2006, 3010–3012 RSC.
- Y. Zhang, P. C. Zhu and D. Edgren, J. Polym. Res., 2010, 17, 725–730 CrossRef CAS.
- Y. H. Wang and Y. L. Hsieh, J. Appl. Polym. Sci., 2010, 116, 3249–3255 CrossRef CAS.
- P. A. Ramires and E. Milella, J. Mater. Sci.: Mater. Med., 2002, 13, 119–123 CrossRef CAS.
- W. N. E. van Dijk-Wolthuis, O. Franssen, H. Talsma, M. J. van Steenbergen, J. J. Kettenes-van den Bosch and W. E. Hennink, Macromolecules, 1995, 28, 6317–6322 CrossRef CAS.
- A. V. Reis, A. R. Fajardo, I. T. A. Schuquel, M. R. Guilherme, G. J. Vidotti, A. F. Rubira and E. C. Muniz, J. Org. Chem., 2009, 74, 3750–3757 CrossRef CAS.
- S. Xing and G. Zhao, Mater. Lett., 2007, 61, 2040–2044 CrossRef CAS.
- J. R. Zhang, T. Qiu, H. F. Yuan, W. Z. Shi and X. Y. Li, Mater. Lett., 2010, 65, 790–792 CrossRef.
- W. H. Eisa, Y. K. Abdel-Moneam, A. A. Shabaka and A. M. Hosam, Spectrochim. Acta, Part A, 2012, 95, 341–346 CrossRef CAS.
- J. Song, H. Kang, C. Lee, S. H. Hwang and J. Jang, ACS Appl. Mater. Interfaces, 2012, 4, 460–465 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of PVA, dextran and methyl cellulose and their corresponding methacrylated derivatives. TGA analysis of PVA, dextran and methyl cellulose nanofibers, with and without the presence of Ag-nanoparticles. Photograph images showing the antimicrobial activity against E. coli growth with PVA–Ag nanofibers. See DOI: 10.1039/c2ra20637d |
| This journal is © The Royal Society of Chemistry 2012 |