DOI:
10.1039/C2RA20359F
(Paper)
RSC Adv., 2012,
2, 6831-6837
Novel Cu–Fe bimetal catalyst for the formation of dimethyl carbonate from carbon dioxide and methanol†
Received
28th February 2012
, Accepted 17th May 2012
First published on 22nd May 2012
Abstract
A novel Cu–Fe bimetal supported catalytic system was prepared and applied to the direct dimethyl carbonate (DMC) formation from methanol and CO2. The prepared catalysts were characterized by means of temperature-programmed reduction (TPR), X-ray powder diffraction (XRD), laser Raman spectra (LRS), X-ray photoelectron spectroscopy (XPS) and temperature programmed desorption (TPD). Metallic Cu, Fe and oxygen deficient Fe2O3−x (0 < x < 3) were formed during the reduction and activation step. The supported Cu–Fe bimetal catalysts exhibited good catalytic activity and high stability for the direct DMC formation. Under the reaction conditions at 120 °C and 1.2 MPa with space velocity of 360 h−1, the highest methanol conversion of 5.37% with DMC selectivity of 85.9% could be achieved. The high catalytic performance of the Cu–Fe bimetal catalysts in the DMC formation could be attributed to the interaction of base sites functioned by metallic Cu and Fe with acid sites provided by oxygen deficient Fe2O3−x (0 < x < 3) in the activation of methanol and CO2. The moderate concentration balance of acid and base sites was in favor of DMC formation.
Introduction
Dimethyl carbonate (DMC) has attracted much attention as a green chemical that can replace phosgene and dimethyl sulfate in the reaction of carbonylation and methylation.1 It is also a promising octane enhancer due to its high oxygen content.2 Several routes have been developed for DMC synthesis.3 Among these DMC synthesis technologies, the straightforward reaction of CO2 with methanol is the most preferred route as it adheres to green chemistry principles.4 Moreover, conversion of CO2 into useful industrial compounds5 has attracted much attention for its environmentally benign nature (i.e., nontoxic, noncorrosive, and nonflammable). The activation of CO2 is the key problem in its conversion to DMC.6 Currently, various catalysts for this reaction have been developed, such as organometallic compounds,7 metal(IV) tetra-alkoxide,8 potassium carbonate,9 zirconia-based10–12 catalysts, cerium-based13–15 catalysts, heteropoly compounds,16 H3PO4–V2O5,17 Cu–Ni bimetallic18–21 supported on different carriers. Among those catalysts available for the direct DMC formation, copper-based catalysts such as the graphite oxide supported Cu–Ni bimetallic particles were considered as one of the most promising catalysts.20 However, it is still far from satisfactory for the scale-up industrial application due to the sintering properties of active components, poor stabilization of final catalysts and tedious procedures of catalyst preparation from a practical point of view. Many factors influence the catalytic performances of supported metal catalysts, such as the metal particle size, metal dispersion, morphology of metal nanoclusters, the character of metal–support interactions and electronic properties of the metal.22 The sintering properties could agglomerate active components into larger particles, which may not be uniformly dispersed on the surface of the catalyst support. The uneven distribution of active components leads to negative effects on the catalytic performance.
The knowledge about reaction mechanisms and information about the role of different types of active sites in a given catalytic reaction is the key factor in designing effective supported catalysts. Spivey, et al.23 nicely gave the details about the relationships between the catalytic performances and active sites of heterogeneous catalysis in catalytic reactions: group VII and IB transition metal elements (Pt,Pd,Ru,Ni,Cu,Co and Au,Ag) can easily be reduced to zero valence for their low heat formation of metal oxides; metal oxides with high heat of formation (e.g. SiO2,TiO2,ZrO2,Al2O3,CeO2) are ordinarily applied as catalyst supports because they are difficult to be reduced; other transition metals such as V,Cr,Mo,W,Mn,Fe,Zn,Re, and sp-metals such as Ga, In, Ge and Sn with an intermediate value as the heat of formation of their oxides are usually present as promoters in supported metal catalysts in the form of isolated ions or nano-sized oxide crystallites even under reducing or reaction conditions. These metal ions behave as Lewis acid sites. As reported in the literature,24–26 the acid–base properties of catalysts play significant roles in determining the catalytic performance in the direct DMC formation from CO2 and methanol, and the amount of DMC produced over the bifunctional catalysts increases by increasing both acidity and basicity of the catalysts. It is reasonable to design bifunctional catalysts for this reaction. Metal nanoclusters of Group VII and IB supported and stabilized on irreducible oxides and promoted by a metal ion can fulfill this requirement. The introduction of inexpensive Fe to the Cu-based catalysts for various organic syntheses has been studied in many papers.27–30 The addition of Fe could prevent the sintering of Cu and also inhibit the oxidation of copper surfaces during the reaction. Furthermore, the introduction of Fe into the Cu-based catalyst can promote the catalytic activity due to the strong interaction between Cu and Fe. Although the studies on Cu–Fe based catalysts are extensive, very little information of the investigation on the DMC formation over Cu–Fe bimetal catalysts is disclosed. In this work, the supported Cu–Fe bimetal catalysts were prepared by the citric-acid complex method31 under simple synthetic conditions and applied to the direct DMC formation from CO2 and methanol. The effects of reduction temperature of the Cu–Fe clusters on catalytic performance was discussed with an aim to establish a fundamental understanding of the properties of the catalyst. A recycle test of the catalyst was also conducted. In addition, we investigated the relation between the catalytic activities and surface acid–base properties of the catalysts. It is found that moderate ratios of acid and base sites are important for the direct DMC formation.
Experimental
An aqueous solution of citric acid and the metal precursors, such as iron nitrate, copper nitrate, were stirred for 2 h. The solution was then impregnated over silica (China GBS High-tech &Industry Co. Ltd., HL-380). The mixture was stirred and aged for 24 h at room temperature, followed by keeping at 60 °C for 1 h until it became gelatinous. The as-prepared samples were dried at 120 °C overnight before being calcined at 550 °C in air for 4 h to give the catalyst precursors. All catalysts were reduced in a H2 stream at desired temperatures for 4 h prior to each catalytic measurement.
Catalyst characterization
The specific surface areas (SBET, m2g−1) of samples were estimated with the N2 adsorption BET method at the liquid nitrogen temperature on the Quantachrom Chem-BET 3000. Temperature programmed reduction (TPR) profiles of catalyst precursors were collected on the Quantachrom Chem-BET 3000 apparatus equipped with a thermal conductivity detector (TCD). 50 mg samples placed in a quartz U-shaped tube were reduced in 5%H2/N2 flow (80 mL min−1) from room temperature to 700 °C at a temperature rise of 5 °C min−1. Samples were pretreated in N2 flow (50 mL min−1) at 300 °C for 2 h to remove the adsorbed water and other contaminants prior to each TPR experiment. X-Ray diffraction (XRD) patterns of samples were recorded on the Rigaku D-Max 2200 diffractometer using graphite-filtered Cu Ka radiation (λ = 0.154178 nm) at 40 kV and 30 mA with a scanning rate of 4° min−1 from 2θ = 30° to 2θ = 80°. The crystallite sizes of the particles were estimated from the XRD peak widths using the Scherrer equation. The morphologies and microstructure of SiO2 and metal particles were examined using a scanning electron microscope (JSM-5600LV system of JEOL) equipped with an energy dispersive X-ray detector (EDX). The accelerating voltage was 15 kV. The laser Raman spectra (LRS) of catalysts was obtained on a laser Raman apparatus (Renoshaw inVia) with the 785 nm line of an Ar+ laser as the excitation source. X-Ray photoelectron spectrum (XPS) of samples was obtained using an ESCALAB250 (Thermo-VG Scientific) spectrometer, equipped with a monochromatised Al Ka (1486.6 eV, 15 kV, and 150 W) source. XPS spectra were recorded with pass energy 20 eV, and the C1s line (284.6 eV) was taken as a reference to correct for electrostatic charging. Temperature programmed desorption (TPD) measurements were performed on a Quantachrom Chem-BET 3000 apparatus. About 100 mg catalyst precursor was first reduced in a 5%H2/N2 flow (30 mLmin−1) at the desired temperatures for 30 min and cooled to 50 °C, followed by exposing to a stream of adsorbed gas (pure CO2 or NH3, 50 mLmin−1) for 1 h, and then the degassing process was performed with a heating rate of 8 °C min−1 under a Helium flow (30 mL min−1), CO2 or NH3 desorbed was detected by the TCD.
The direct DMC formation from CO2 and methanol was investigated in a continuous flow system with a fixed-bed micro-reactor (Scheme 1 and 2, see ESI†) . 2 g fresh catalyst (40–50 mesh) was packed in the tubular reactor. Pure N2 was fed into the reactor to exhaust the air inside before the catalyst bed was heated to the desired temperature. CO2 controlled by a mass flow controller was used together with methanol, which was introduced by passage of the CO2 carrier gas through a saturator maintained at 35 °C. CO2 and methanol-saturated CO2 streams were heated in a pre-heater before it was passed into the reactor to afford the experimental pressure. The catalytic reactions were carried out at temperatures of 90–140 °C, under pressures of 0.3–1.5 MPa and space velocities of 360–1080 h−1 with time on stream for 7 h. The gaseous products' stream was introduced to the gas chromatograph (GC7890F) equipped with a flame ionization detector (FID) through an on-line six-way valve. The condensed liquid collected by the cooling separater was analyzed by the gas chromatograph mass spectrometer (GCMS-QP2010 plus) to confirm the DMC formation in this reaction. The main reaction products of CO2 reacting with methanol over Cu–Fe/SiO2 were DMC and H2O, and there were a few by-products of CH2O, CO and dimethyl ether (DME). Catalytic performances were indicated as methanol conversion (Xc %), DMC selectivity (SDMC %), and DMC yield (yield %). These parameters were calculated by the following equations:| | 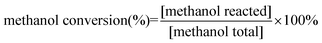 | (1) |
| | 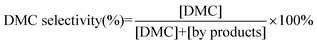 | (2) |
| | | DMC yield(%) = methanol conversion × DMC selectivity × 100% | (3) |
Results and discussion
Catalytic performance
The catalytic performances of direct DMC formation from CO2 and methanol over silica supported Cu–Fe bimetal catalysts with varying molar ratios of metals and total metal contents are depicted in Table S1 (see ESI†). It appears that there exists an optimum composition for attaining maximum activity, and this composition corresponds to 15% (3CuO–Fe2O3)/SiO2, and the synergetic effect of Cu and Fe clusters in the activation of methanol and CO2 is the key factor in dominating catalytic performance. These results could be confirmed by the following experiments. Therefore, a total loading of metal CuO + Fe2O3 = 15 wt% and the metal molar ratio of Cu/Fe = 3/2 loaded on SiO2 (3Cu–2Fe/SiO2) were further used as the optimal Cu–Fe bimetal catalytic system to obtain the fundamental properties of the catalyst in the direct DMC formation. In this work, the effects of reaction conditions on the catalytic performances of DMC formation over the 3Cu–2Fe/SiO2 catalysts are carried out, and the results are summarized in Table S2 (see ESI†). The catalytic performances go through a peak value at 120 °C in the reaction temperature range from 90 to 140 °C. Higher reaction pressures favor the direct DMC formation. The catalyst activities decline marginally with increasing space velocity. Hence, the reaction conditions at 120 °C and 1.2 MPa with a space velocity of 360 h−1 are selected for further exploration.
The effects of reduction temperatures on the catalytic performance of DMC formation over 3Cu–2Fe/SiO2 at a fixed reaction time of 2.5 h were studied, and the results are shown in Fig. 1. It is evident that the methanol conversion first increases with increasing reduction temperature, and then it drops rapidly with a further increase in the reduction temperature to 650 °C, while the DMC selectivity showed an upwards trend with increasing reduction temperature. The highest methanol conversion of 5.37% with DMC selectivity of 85.9% can be obtained when the reduction temperature reached 600 °C. Obviously, the reduction processes of the 3Cu–2Fe/SiO2 precursor lead to great changes in catalytic activities, indicating that the catalyst reduced at various temperatures undergoes several structure transformations.
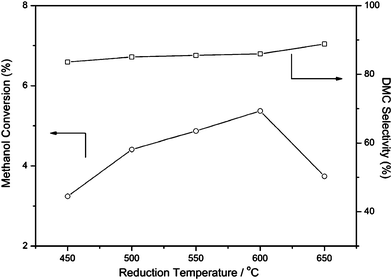 |
| | Fig. 1 The influences of reduction temperatures on the catalytic performance of DMC formation over 3Cu–2Fe/SiO2 at 2.5 h of the reaction. Reaction conditions: temperature 120 °C; pressure 1.2 MPa; space velocity 360 h−1; catalyst weight 2 g; time on stream 7 h. | |
Moreover, the results shown in Fig. 2 demonstrate that there is a similar trend of the catalytic performance over all catalysts reduced at various temperatures with time on stream. The methanol conversions increase within 2.5 h from the start of the reaction, which could be attributed to the generation of abundant active species of reactants in the initial reaction stage. Meanwhile, the DMC selectivity decrease with reaction time. The decreasing trend of catalytic performance over the period from 3–7 h indicates the continuous deactivation of catalysts. Presumably, the deactivation of catalysts is due to the change of chemical states of active components during the reaction. The change of the chemical situation for active components can be confirmed by LRS results (Fig. S1, see ESI†) of the catalysts before and after the catalytic reaction. In order to further explore the stabilities of the 3Cu–2Fe/SiO2 for this reaction, an attempt was made to recover the used catalysts by re-calcinations and re-reduction and to reuse it for subsequent run under the same reaction conditions. The catalytic performance and SBET of the catalyst being recycled are presented in Table 1. The catalysts are found to be active up to three consecutive cycles. Both the catalytic activities and SBET slightly drop with catalytic run numbers. This may be due to the re-dispersion of active sites and sintering of Cu–Fe clusters during the regeneration of catalyst. However, the 3Cu–2Fe/SiO2 exhibits fairly good durability for the direct DMC formation from CO2 and methanol.
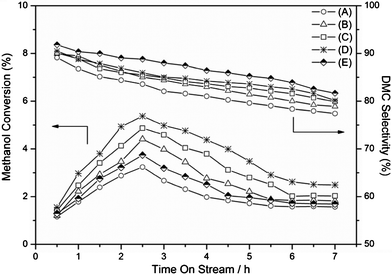 |
| | Fig. 2 Catalytic performance of 3Cu–2Fe/SiO2 reduced at various temperatures with time on stream (A) 450 °C, (B) 500 °C, (C) 550 °C, (D) 600 °C, (E) 650 °C, reaction conditions: temperature 120 °C; pressure 1.2 MPa; space velocity 360 h−1; catalyst weight 2 g; time on stream: 7 h. | |
Table 1 Recyclability study of Cu–Fe/SiO2 catalyst for the direct DMC formation from CO2 and methanola
| Entry |
Recycle number |
Methanol conversion (%) |
DMC selectivity (%) |
DMC yield (%) |
S
BET (m2 g−1) |
|
The Cu–Fe/SiO2 catalyst was reduced in a H2 stream under 600 °C for 4 h. Reaction conditions: temperature 120 °C; pressure 1.2 MPa; space velocity 360 h−1; catalyst weight: 2 g; time on stream: 7 h. The methanol conversion was the average data between 2–4 h with time on stream.
|
| 1 |
Fresh |
4.98 |
84.5 |
4.20 |
248.6 |
| 2 |
First |
4.64 |
80.8 |
3.75 |
232.4 |
| 3 |
Second |
4.32 |
77.5 |
3.35 |
215.1 |
| 4 |
Third |
3.91 |
73.8 |
2.86 |
198.7 |
Characterizations of catalysts
The TPR profiles of Cu/SiO2, Fe/SiO2 and 3Cu–2Fe/SiO2 catalyst precursors are depicted in Fig. 3. As shown in Fig. 3(A), two H2 consumption peaks appear around 220 °C and 260 °C, which are assigned to the reduction of well dispersed CuO and bulk CuO,19 respectively. The peaks in the temperature range 280–500 °C in Fig. 3(B) are ascribed to the reduction of Fe2O3 to Fe3O4, probably subsequent to FeO, and then it is totally reduced to metallic Fe above 670 °C.27 According to these results, the first reduction temperature observed at the 200–300 °C region in Fig. 3(C) is due to the reduction of CuO to Cu, the second broad reduction peak at 350–500 °C is associated with the reduction of Fe2O3 to Fe3O4 or FeO, and the temperature region around 650 °C is responsible for the complete reduction of iron oxide to metallic Fe.
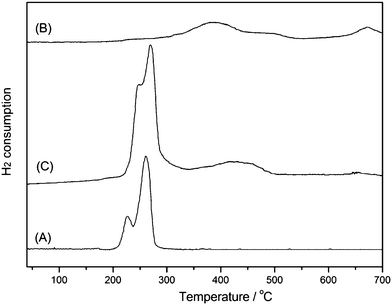 |
| | Fig. 3 TPR profiles of catalyst precursors (A) Cu/SiO2, (B) Fe/SiO2, (C) 3Cu–2Fe/SiO2. | |
XRD patterns of the catalyst precursor and catalysts after reductions in H2 stream at various temperatures are depicted in Fig. 4. The catalyst precursor (Fig. 4 (A)) showed highly crystallized monoclinic CuO and rhombohedral a-Fe2O3. In the XRD spectra of all catalysts treated in the H2 stream under temperatures above 450 °C (Fig. 4(B)–(F)), it is observed that the copper oxide species are entirely reduced to metallic Cu, whereas iron oxide cannot be reduced to metallic Fe under the reduction temperatures below 600 °C. There is no FeO diffraction peak observed because FeO is a metastable species and easily oxidized. When the reduction temperature increased to 650 °C, the iron oxide species can be fully reduced to metallic Fe. Generally speaking, it is difficult for H2 to reduce Fe2O3 from trivalence to zero valences at lower reduction temperatures. However, its chemical state would change to a certain extent after being treated by H2. For the 3Cu–2Fe/SiO2 precursor reduced at temperatures between 450 °C and 650 °C, iron element exists in a certain middle valence state forming oxygen unsaturated Fe2O3−x (0 < x < 3), and higher reduction temperatures lead to lower Fen+(0 < n < 3) content. The changes in XRD patterns in a series of reduction treatments are unambiguously interpreted with the TPR profiles for the 3Cu–2Fe/SiO2 precursor. The particle size of metallic Cu estimated from the Scherrer equation using a (111) (2θ = 43.24°) diffraction plane increases with increasing reduction temperature: 19.5 nm, 21.1 nm, 22.5 nm 25.0 nm, 29.7 nm for the catalyst reduced at 450 °C, 500 °C, 550 °C, 600 °C, 650 °C. It seems that oxygen unsaturated Fe2O3−x (0 < x < 3) is indispensable in preventing metallic Cu from sintering. From the SEM images of the 3Cu–2Fe/SiO2 catalyst reduced at 600 °C (Fig. S2(B), ESI†), it is evident that active metal particles (white circular dot) are evenly dispersed on the surface of SiO2. The energy dispersive X-ray detector (EDX) (Fig. S2(C), ESI†) suggests that these white circular dots include Cu,Fe elements.
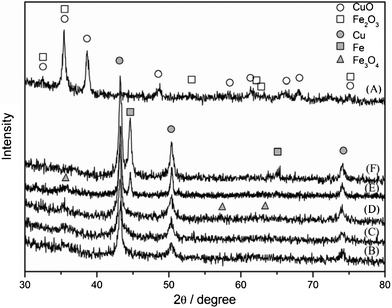 |
| | Fig. 4 XRD patterns of (A) 3Cu–2Fe/SiO2 precursor and (A) reduced at (B) 450 °C, (C) 500 °C, (D) 550 °C, (E) 600 °C, (F) 650 °C. | |
The XPS measurements of 3Cu–2Fe/SiO2 precursor reduced at 600 °C were performed to explore the oxidation states of Cu and Fe and the interactions of active components with SiO2. As shown in Fig. 5, the binding energy of Cu (2p3/2) for the catalysts centered at 933.10 eV, accompanied by a weak satellite peak, is attributed to Cu (0). Such a BE value is slightly different from those reported in the literature.32 This indicates that the chemical environment of the Cu changes, presumably owing to the interaction of Cu with Fe species or SiO2 during the activating process. For the survey scan of Fe (2p3/2) presented in Fig. 6, the binding energy observed at 710.47 eV is smaller than that of Fe3O4 at 710.8 eV33 and closer to that of FeO at 710.3.34 The presence of the weak shoulder satellite peak is consistent with the results obtained by other researchers,35 and is clear evidence of the existence of Fe2+. An inconspicuous peak observed at 706.94 eV implies the presence of Fe(0) on the surface. These data on the XPS spectra suggest that the 3Cu–2Fe/SiO2 precursor is reduced at 600 °C results in metallic Cu, Fe and oxygen unsaturated Fe (Fe2O3−x, 0 < x < 3). These are in accordance with the results of TPR (Fig. 3) and XRD (Fig. 4).
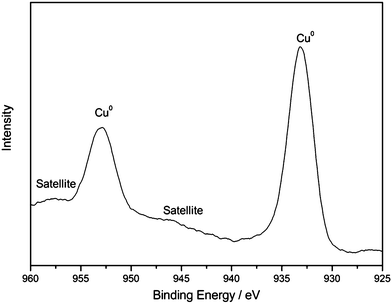 |
| | Fig. 5 XPS pattern of Cu2p for the 3Cu–2Fe/SiO2 precursor reduced at 600 °C. | |
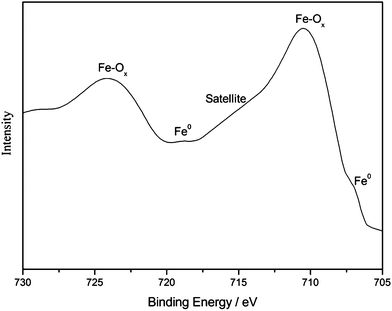 |
| | Fig. 6 XPS pattern of Fe2p for the 3Cu–2Fe/SiO2 precursor reduced at 600 °C. | |
The acid–base properties of 3Cu–2Fe/SiO2 were characterized by TPD–NH3 and TPD–CO2, and the results are shown in Fig. 7 (TPD–NH3) and Fig. 8 (TPD–CO2), respectively. There is a major desorption peak in the TPD–NH3 (Fig. 7) around 200 °C over all catalysts, which is ascribed to one kind of weak acid site. The amount of NH3 desorbed from 3Cu–2Fe/SiO2 decreases with increasing reduction temperature. This indicates that the number of the acid sites declined at higher reduction temperature. On the other hand, from the profiles of TPD–CO2 depicted in Fig. 8, it is found that the maximum peak temperatures determined by Gaussian deconvolution are 120 °C, 175 °C and 340 °C, corresponding to three types of base sites in catalysts. The desorption areas of the 120 °C peak first increase with reduction temperatures from 450 °C to 600 °C, but it decreases slightly by further increasing the reduction temperature to 650 °C. This suggests that the number of weak base sites runs through a maximum at 600 °C. Relating to the performances of the catalysts as a function of reduction temperatures (Fig. 1) and reaction temperatures (Table S2, ESI†), it is reasonable to assume that the weak base sites favor the DMC formation. The entire desorption area, in the range 100–550 °C, slightly rises with higher reduction temperatures, and this suggests that the amount of base sites increase with increasing reduction temperature. According to the results of TPR, XRD and XPS (Fig. 3, 4, 5 and 6), metallic Cu, Fe and oxygen deficient Fe2O3−x (0 < x < 3) were formed during the reduction process, and a higher reduction temperature was reasonable for the smaller amount of Fen+ (0 < n < 3). Consequently, we can conclude that the acid behavior of the catalyst is mainly the function of oxygen deficient Fe2O3−x (0 < x < 3), while the base behavior is the result of Cu and Fe metals. Under the same reduction conditions, the larger amount of Cu species gives a larger number of base sites, and the higher content of Fe species leads to more acid sites on the catalyst surface.
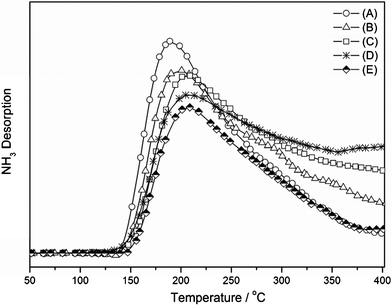 |
| | Fig. 7 TPD–NH3 profiles of the 3Cu–2Fe/SiO2 precursor reduced at (A) 450 °C, (B) 500 °C, (C) 550 °C, (D) 600 °C, (E) 650 °C. | |
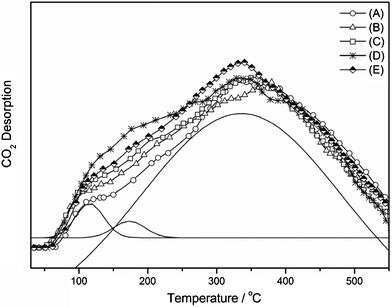 |
| | Fig. 8 TPD–CO2 profiles of the 3Cu–2Fe/SiO2 catalyst precursor reduced at (A) 450 °C, (B) 500 °C, (C) 550 °C, (D) 600 °C, (E) 650 °C. | |
In addition, correlating the catalytic performances as listed in Table S1 (see ESI†) and the acid–base properties investigated by TPD techniques as presented in Fig. S3 and Fig. S4 (see ESI†) of Cu–Fe bimetal catalysts with various Cu/Fe atomic molar ratios, it is found that the changes in catalytic activities do not linearly vary with the concentrations of Cu or Fe, and the best catalytic activity is obtained when Cu/Fe is 3/2, which possesses neither the largest amount of acid nor base sites on the catalyst surface. The bimetal catalysts present better catalytic performances than pure metals. It can be assumed that the iron species, as a promoter, interacts with metallic Cu in the Cu–Fe bimetal catalyst and can form the “metal ion–metal nanocluster” ensemble sites,23 which are responsible for the activation of reactants in this catalytic reaction. The bimetal catalyst with an equal molar ratio of Cu/Fe exhibits the worst catalytic performance, which could be attributed to the lowest concentration of acid and base sites on the catalyst surface. Too many acid sites (Cu/Fe = 2/5) or too many base sites (Cu/Fe = 5/2) in the catalyst presents an inferior catalytic activity compared to the catalyst with suitable acid–base sites (Cu/Fe = 3/2). These results are evidence that a larger amount of acid and base sites, with moderate molar ratios, favors the formation of DMC.
As reported in the literature,24,26 methanol is activated on the acid and base sites of catalysts to afford methyl species and methoxy species, respectively. CO2 adsorbed on the base sites of the catalyst may react with methoxy species to generate methoxy carbonate anions, which may further react with methyl species to produce DMC, that is to say, more acid sites favor the activation of methanol while more base sites are beneficial for the activation of CO2. Both acid and base sites are important for direct DMC formation. If the amount of one kind of active site is much more than the other in the catalyst, only the activation of one reactant is dominant, which seems unfavorable in the DMC yield. In our present work, inexpensive Fe instead of Ni cooperated with Cu loaded on SiO2 is developed as a catalyst for direct DMC formation. It is found that the Cu–Fe exhibited good catalytic activity, although it is not as good as a Cu–Ni bimetal catalyst,18–21 in which the bimetallic Cu–Ni is served as base sites, while the carriers are usually treated by complicated procedures to provide the acid sites. The importance is that this is the first report of Cu–Fe bimetal catalyst used for the formation of DMC. Even though the same Cu–Ni particles were used as active components, the DMC yields were different. The effective activity of the Cu–Fe catalyst for this reaction is ascribed to the interaction of base sites offered by metallic Cu and Fe with acid sites afforded by oxygen deficient Fe2O3−x (0 < x < 3). The iron (3d64s2) naturally has a different electron structure than nickel (3d84s2). There are two electrons less in the 3d-obital of iron than nickel, which indicates that iron would be more easily acted on as an electron acceptor rather than a donor compared with nickel. It is known that the activation of CO2 plays a key role in this reaction; the electron donor would be much more favored in activating CO2. Hence, when the iron or nickel is cooperated with Cu (3d104s1), the nickel exhibits better ability to active the reactants than iron. In addition, the iron species in the catalyst is unstable and easily transformed from lower to higher valence as the reaction proceeds. These might be the reasons that the DMC formation over Cu–Ni bimetal catalysts shows better activity than that of Cu–Fe bimetal catalysts. If it is possible to synthesize a catalyst, which has a large amount of acid and base sites with moderate molar ratios, the DMC yield will be improved drastically.
Conclusions
A series of supported Cu–Fe bimetal catalysts for the DMC formation from CO2 and methanol were developed. The Cu–Fe nanoclusters showed effective and stable catalytic activity in this reaction. The Cu–Fe oxides with a molar ratio of Cu/Fe = 3/2 reduced at 600 °C revealed the best catalytic activity. The catalyst was found to be active for up to three consecutive cycles, and the highest methanol conversion of 5.37% with DMC selectivity of 85.9% can be acquired under reaction conditions at 120 °C and 1.2 MPa with space velocity of 360 h−1. Metallic Cu, Fe and oxygen deficient Fe2O3−x (0 < x < 3) were formed in the reduction and activation step. The effective activity of the Cu–Fe bimetal catalyst for this reaction was ascribed to the interaction of base sites offered by metallic Cu and Fe with acid sites afforded by oxygen deficient Fe2O3−x (0 < x < 3) in activating methanol and CO2. Both acid and base sites are important, and a suitable concentration balance of acid and base sites favors the DMC formation.
Acknowledgements
The authors would like to thank the China High-Tech Development 863 Program (2009AA034900, 2009AA03Z340), Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (2010), Guangdong Province Sci & Tech Bureau (Key Strategic Project Grant No. 2008A080800024, 10151027501000096), and Chinese Universities Basic Research Founding for financial support of work. Y. L. thanks National University of Singapore for financial support.
References
- Y. Ono, Appl. Catal., A, 1997, 155, 133–166 CrossRef CAS.
- M. A.Pacheco and C. L. Marshall, Energy Fuels, 1997, 11, 2–29 CrossRef.
- M. Aresta, Quim. Nova, 1999, 22, 269–272 CrossRef CAS.
- D. Delledonne, F. Rivetti and U. Romano, Appl. Catal., A, 2001, 221, 241–251 CrossRef CAS.
- I. Omae, Catal. Today, 2006, 115, 33–52 CrossRef CAS.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- J. Kizlink, Collect. Czech. Chem. Commun., 1993, 58, 1399–1402 CrossRef CAS.
- J. Kizlink and I. Pastucha, Collect. Czech. Chem. Commun., 1995, 60, 687–692 CrossRef CAS.
- S. Fang and K. Fujimoto, Appl. Catal., A, 1996, 142, L1–L3 CrossRef CAS.
- K. Tomishige, T. Sakaihori, Y. Ikeda and K. Fujimoto, Catal. Lett., 1999, 58, 225–229 CrossRef CAS.
- Y. Ikeda, T. Sakaihori, K. Tomishige and K. Fujimoto, Catal. Lett., 2000, 66, 59–62 CrossRef CAS.
- K. Tomishige, Y. Furusawa, Y. Ikeda, M. Asadullah and K. Fujimoto, Catal. Lett., 2001, 76, 71–74 CrossRef CAS.
- J. C. Choi, L. N. He, H. Yasuda and T. Sakakura, Green Chem., 2002, 4, 230–234 RSC.
- M. Aresta, A. Dibenedetto, C. Pastore, C. B. Corrado, A. B. Brunella, S. Cometa and E. De Giglio, Catal. Today, 2008, 137, 125–131 CrossRef CAS.
- M. Aresta, A. Dibenedetto, C. Pastore, A. Angelini, B. Aresta and I. Papai, J. Catal., 2010, 269, 44–52 CrossRef CAS.
- L. A. Allaoui and A. Aouissi, J. Mol. Catal. A: Chem., 2006, 259, 281–285 CrossRef CAS.
- X. L. Wu, M. Xiao, Y. Z. Meng and Y. X. Lu, J. Mol. Catal. A: Chem., 2005, 238, 158–162 CrossRef CAS.
- X. L. Wu, Y. Z. Meng, M. Xiao and Y. X. Lu, J. Mol. Catal. A: Chem., 2006, 249, 93–97 CrossRef CAS.
- X. J. Wang, M. Xiao, S. J. Wang, Y. X. Lu and Y. Z. Meng, J. Mol. Catal. A: Chem., 2007, 278, 92–96 CrossRef CAS.
- J. Bian, M. Xiao, S. J. Wang, X. J. Wang, Y. X. Lu and Y. Z. Meng, Chem. Eng. J., 2009, 147, 287–296 CrossRef CAS.
- J. Bian, M. Xiao, S. J. Wang, Y. X. Lu and Y. Z. Meng, Catal. Commun., 2009, 10, 1142–1145 CrossRef CAS.
- A. Y. Stakheev and L. M. Kustov, Appl. Catal., A, 1999, 188, 3–35 CrossRef CAS.
- J. J. Spivey, G. W. Roberts, J. L. Margitfalvi and S. Gőbölös, Catalysis, 2004, 17, 1–104 Search PubMed.
- A. D. M. Aresta, C. Pastore, I. Papai and G. Schubert, Top. Catal., 2006, 40, 71–81 CrossRef.
- S. H. Fujita, B. M. Bhanage, Y. Ikushima and M. Arai, Green Chem., 2001, 3, 87–91 RSC.
- K.W. La, J. C. Jung, H. Kim, S. H. Baeck and I. K. Song, J. Mol. Catal. A: Chem., 2007, 269, 41–45 CrossRef CAS.
- C. S. Chen, W. H. Cheng and S. S. Lin, Appl. Catal., A, 2004, 257, 97–106 CrossRef CAS.
- H. Ando, Q. Xu, M. Fujiwara, Y. Matsumura, M. Tanaka and Y. Souma, Catal. Today, 1998, 45, 229–234 CrossRef CAS.
- M. G. Lin, K. G. Fang, D. B. Li and Y. Sun, Catal. Commun., 2008, 9, 1869–1873 CrossRef CAS.
- S. R. Yan, K. W. Jun, J. S. Hong, M. J. Choi and K.W. Lee, Appl. Catal., A, 2000, 194–195, 63–70 CrossRef CAS.
- K. Faungnawakij, Y. Tanaka, N. Shimoda, T. Fukunaga, R. Kikuchi and K. Eguchi, Appl. Catal., B, 2007, 74, 144–151 CrossRef CAS.
- B. Lerebours, J. Dürr, A. D. Huisser, J. P. Bonnelle and M. Lenglet, Phys. Status Solidi A, 1980, 61, K175–K180 CrossRef CAS.
- D. D. Hawn and B. M. De Koven, Surf. Interface Anal., 1987, 10, 63–74 CrossRef CAS.
- G. C. Allen, M. T. Curtis, A. J. Hooper and P. M. Tucker, J. Chem. Soc., Dalton Trans., 1974, 1525–1530 RSC.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: ESI includes experimental apparatus, the dependence of catalytic performance on the loading and molar ratio of metals and Raman spectra of catalysts before and after being evaluated. See DOI: 10.1039/c2ra20359f/ |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.