Doxorubicin is a photocatalyst for the generation of H2O2†
Krzysztof
Nawara
ab,
Pawel
Krysinski
b and
G. J.
Blanchard
*a
aMichigan State University, Department of Chemistry, East Lansing, MI 48824, USA. E-mail: blanchard@chemistry.msu.edu; Tel: +1 517-355-9715 x224
bUniversity of Warsaw, Department of Chemistry, Pasteura 1, 02-093, Warsaw, Poland
First published on 9th March 2012
Abstract
We have investigated the photoreactivity of the chemotherapeutic agent doxorubicin. We demonstrate the ability of this compound to generate H2O2 catalytically as a result of auto-oxidation of its photoreduced form. H2O2 may accumulate in toxic levels in pharmaceutical preparations.
Cardiotoxicity is the major adverse effect that limits the effectiveness of anthracyclines in anticancer therapy.1 Doxorubicin (Adriamycin™), one member of the anthracycline family of pharmaceuticals, has been used for over fifty years to treat a variety of cancers. The probability of developing chemically-induced heart failure is correlated with the total dose of doxorubicin administered. A retrospective study revealed that the probability of developing congestive heart failure is 3% for a 400 mg m−2 dose, 7% for 550 mg m−2 and 18% for 700 mg m−2.2 A maximum dose of 550 mg m−2 for doxorubicin was suggested to minimize the risk of heart failure.3 It has also been reported that the only successful approach to the treatment of doxorubicin-induced heart failure is heart transplantation.4 Because the rate of cardiomyocyte replication in humans is comparatively low,5 it is unlikely that the cardiotoxicity of anthracyclines is related to the inhibition of the cell-division cycle, the mechanism of anticancer activity for this family of compounds. The mechanism of anthracycline cardiotoxicity has been considered at length in review articles by Jung and Reszka,6 and Minotti et al.7 Researchers have linked the formation of reactive oxygen species (ROS) such as the superoxide anion, hydrogen peroxide and hydroxyl radical, species that cause lipid peroxidation,8 oxidative DNA damage9 and cardiomyocyte hypertrophy,10 with anthracycline cardiotoxicity. This finding is predicated on the fact that the human heart is poorly equipped with detoxifying enzymes relative to other tissues.11
The generation of reactive oxygen species occurs in two sequential reactions known collectively as anthracycline redox cycling.12 In the first reaction, anthracyclines undergo a one electron enzymatic reduction to form the corresponding semiquinone radical. This step can be catalysed by various enzymes including cytochrome P450 or -b5 reductases, mitochondrial NADH dehydrogenase, xanthine dehydrogenase and endothelial nitric acid synthase.7,12 Subsequently, the spontaneous oxidation of the semiquinone radical with dissolved oxygen occurs, regenerating the anthracycline and forming superoxide anion, hydrogen peroxide and hydroxyl radicals. The rate of ˙OH radical generation can be enhanced by the presence of Fe2+ and/or Fe3+ through the iron-catalysed Haber–Weiss reaction,13 and haemoglobin is also known to facilitate the formation of ˙OH.14
The reduction of anthracyclines can proceed enzymatically as well as by other means. Anthraquinone photoreduction is well known15,16 and has been discussed previously in the context of simple anthraquinones17 and α-hydroxyanthraquinones18 dissolved in organic solvents. In the work presented here, we demonstrate that doxorubicin undergoes photoinduced reduction to the corresponding dihydroquinone in water, with subsequent spontaneous oxidation of the photoproduct back to doxorubicin with the formation of hydrogen peroxide (Scheme 1).
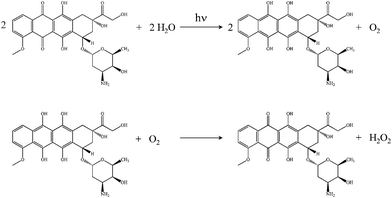 | ||
| Scheme 1 Reactions of doxorubicin. Top: Doxorubicin photoreduction to form the corresponding dihydroquinone and O2. Bottom: Oxidation of the dihydroquinone to form doxorubicin and H2O2. | ||
These reactions provide a possible explanation for the chemically induced cardiotoxic side effects exhibited by doxorubicin. These findings are also consistent with the fact that doxorubicin encapsulated in polyethyleneglycol-coated liposomes (DOXIL™) displays much lower cardiotoxicity than doxorubicin in solution;19 PEG absorbs UV-A, thus attenuating photoinduced reduction and degradation of doxorubicin.20
Lown and Chen21 discovered that chemically reduced doxorubicin gave rise to free hydroxyl radicals. They indicated that the rate of ˙OH formation is similar to the rate of ˙OH production seen in dilute H2O2 solutions. To evaluate whether their findings are consistent with the catalytic production of H2O2, we measured the chemical reduction of doxorubicin (Fig. 1). The introduction of NaBH4 to a doxorubicin solution markedly decreases doxorubicin fluorescence intensity, consistent with doxorubicin reduction. However, after reaching a minimum (corresponding to the consumption of BH4−), the doxorubicin fluorescence intensity slowly increases due to spontaneous oxidation of the reduced species mediated by dissolved molecular oxygen in solution.
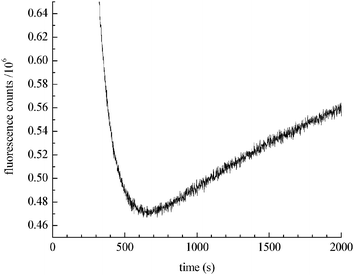 | ||
| Fig. 1 Kinetics of doxorubicin reduction by NaBH4. The decrease in fluorescence intensity at 590 nm is due to reduction of doxorubicin. After 600 s the rate of auto-oxidation exceeds the rate of reduction and doxorubicin regeneration is seen. | ||
The photochemical degradation of doxorubicin has been studied extensively,22–24 but its ability to form H2O2 catalytically has not been examined to date. A comparison of doxorubicin photodegradation time profiles for solutions exposed to air and purged with N2 demonstrates that the net rate of photodegradation is oxygen-dependent (Fig. 2a). The data shown in Fig. 2a are equivalent to the early-time decay data shown in Fig. 1. The recovery seen in Fig. 1 is qualitatively the same as that shown in Fig. 2b. The fact that photodegradation is more facile in the absence of O2 implies that either the reaction kinetics are attenuated by the presence of oxygen or the photoproduct reacts with O2 to regenerate doxorubicin. To resolve this question, we have monitored the fluorescence intensity of doxorubicin following photodegradation, during subsequent exposure of the solution to air. Doxorubicin emission is seen for excitation at both 320 nm and 530 nm, but photodegradation occurs only for 320 nm excitation. Upon exposure of a N2-purged and UV-irradiated sample to air, we observed growth in doxorubicin fluorescence intensity at 590 nm (Fig. 2b). While other photoproducts or oxidation products can, in principle, emit at 590 nm, spectral data reveal no additional bands and only the re-growth of the doxorubicin emission spectrum. This finding indicates that doxorubicin is regenerated spontaneously by exposing the photoproduct to O2. After an initially slow induction period, rapid growth in 590 nm fluorescence intensity occurs until the photoproduct is consumed. The functional form of these data is consistent with a 3/2 order kinetic equation, characteristic of a free radical reaction.25,26 For this reaction we recover a constant k = 6.2 × 10−6 s−1 M−0.5 (±5%).
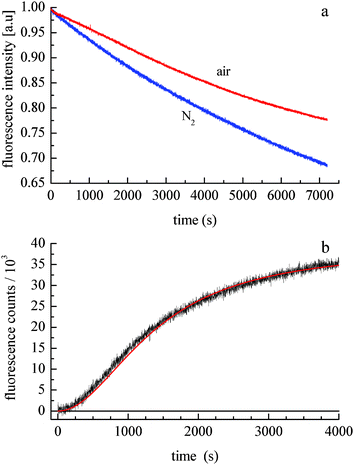 | ||
| Fig. 2 (a) Doxorubicin emission at 590 nm as a function of 320 nm irradiation time. The solution is stirred and purged with N2 (lower trace) or with air (top trace). (b) Doxorubicin 590 nm emission intensity recovery as a function of time following exposure of a N2-purged solution to air. The solid line is the fit of a 3/2 order kinetic equation to the data. | ||
It is useful to differentiate photocatalytic and photoreactive behavior for doxorubicin. Both processes occur, with the former leading to recovery of doxorubicin and formation of H2O2 (Scheme 1), characterized by very different rates for the photoreduction and auto-oxidation reactions. Photoreactivity (not shown) is a different, irreversible reaction that does not lead to recovery of doxorubicin in the absence of UV excitation. The data in Fig. 2b, showing recovery of doxorubicin in the absence of UV excitation, demonstrate the operation of both reactions shown in Scheme 1 and are correlated with the formation of H2O2.
In addition to characterizing the photocatalytic behavior of doxorubicin, it is important to determine the amount of reaction product formed (H2O2, Scheme 1). The detection of hydrogen peroxide in UV-irradiated doxorubicin solutions requires sub-micromolar sensitivity because of the concentrations of doxorubicin we have used, and the detection methodology should operate in a spectroscopic or electrochemical window free from interference by doxorubicin. We use Ampliflu™ Red (10-acetyl-3,7-dihydroxyphenoxazine, known also as Amplex® Red) for the detection of H2O2.27,28 This is a well established, highly sensitive assay that does not suffer from emission spectral interference with doxorubicin. Ampliflu™ Red is a colorless, non-fluorescent compound that reacts with H2O2 in the presence of horseradish peroxidase to form resorufin, a fluorescent compound characterized by an absorption maximum at 571 nm (εmax = 69![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 L mol−1 cm−1),29 and an emission maximum at 587 nm (Φfl = 0.23).30 The emission spectra of resorufin and doxorubicin are overlapped, but their absorption bands are not. Irradiation at 575 nm allows for the selective excitation of resorufin. Because Ampliflu™ Red is photosensitive, we could not follow the kinetics of H2O2 formation during irradiation of the doxorubicin solution. Following 320 nm irradiation of the doxorubicin solution, we introduced Ampliflu™ Red with horseradish peroxidase into the cuvette. The resulting resorufin emission intensity was compared with the emission intensity of a sample that had not been irradiated (Fig. 3a), showing unambiguously the generation of hydrogen peroxide upon 320 nm irradiation of an aqueous doxorubicin solution. The presence of resorufin was also confirmed by absorbance (Fig. 3b). We assert that the presence of resorufin in the non-irradiated sample is due to hydrogen peroxide formed during sample preparation under room light conditions.
700 L mol−1 cm−1),29 and an emission maximum at 587 nm (Φfl = 0.23).30 The emission spectra of resorufin and doxorubicin are overlapped, but their absorption bands are not. Irradiation at 575 nm allows for the selective excitation of resorufin. Because Ampliflu™ Red is photosensitive, we could not follow the kinetics of H2O2 formation during irradiation of the doxorubicin solution. Following 320 nm irradiation of the doxorubicin solution, we introduced Ampliflu™ Red with horseradish peroxidase into the cuvette. The resulting resorufin emission intensity was compared with the emission intensity of a sample that had not been irradiated (Fig. 3a), showing unambiguously the generation of hydrogen peroxide upon 320 nm irradiation of an aqueous doxorubicin solution. The presence of resorufin was also confirmed by absorbance (Fig. 3b). We assert that the presence of resorufin in the non-irradiated sample is due to hydrogen peroxide formed during sample preparation under room light conditions.
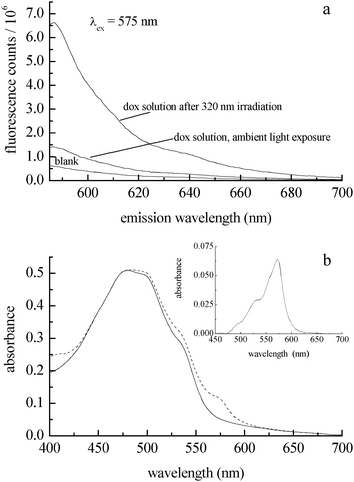 | ||
| Fig. 3 (a) Emission spectrum of resorufin formed from Ampliflu™ Red and H2O2 with horseradish peroxidase (top trace) as a function of irradiation of a 40 μM doxorubicin solution with 320 nm light for 40 min. The middle trace is a doxorubicin solution with Ampliflu™ Red/HRP added, prior to UV irradiation. The bottom trace is a doxorubicin solution without Ampliflu™ Red present. (b) Absorbance spectra of a N2-purged 50 μM doxorubicin solution after 120 min. UV exposure (solid trace). Absorbance of doxorubicin and resorufin (maximum at 571 nm) after exposure to air and introduction of Ampliflu™ Red/HRP (dashed trace). Inset: Difference spectrum, showing the resorufin absorption band, which corresponds to 1 μM H2O2. There is no contribution from doxorubicin emission when resorufin is excited at 575 nm. | ||
In conclusion, the data we report here show that doxorubicin undergoes photoreduction upon UV irradiation to form a dihydroquinone in aqueous solution. Subsequent spontaneous oxidation of the dihydroquinone occurs to regenerate doxorubicin with hydrogen peroxide as a product of that reaction. Kinetic data on the recovery of doxorubicin emission intensity are consistent with a free radical reaction mechanism, analogous to the process of enzymatic redox cycling. While other photodegradation pathways exist for doxorubicin, the dominant reactivity seen for this molecule is the photocatalytic formation of H2O2. Our data offer an explanation for the cardiotoxicity of doxorubicin and the reduced cardiotoxicity of DOXIL™, a formulation that shows higher resistance to UV-A degradation due to the presence of PEG. Further work is underway to provide a detailed mechanistic explanation of the process we report here.
Acknowledgements
This work was supported by Grant CHE 0808677 from the National Science Foundation. KN gratefully acknowledges the support of the Foundation of Polish Science MPD Program co-financed by the European Regional Development Fund.References
- M. Ewer and S. Ewer, Nat. Rev. Cardiol., 2010, 7, 564–575 CrossRef
.
- D. D. Von Hoff, M. W. Layard, P. Basa, H. L. J. Davis, A. L. Von Hoff, M. Rozencweig and F. M. Muggia, Ann. Intern. Med., 1979, 91, 710–717 CAS
- E. A. LeFrak, J. Pitha, S. Rosenheim and S. A. Gottleib, Cancer, 1973, 32, 302–314 CrossRef CAS
- P. K. Singal and N. Iliskovic, N. Engl. J. Med., 1998, 339, 900–905 CrossRef CAS
- O. Bergmann, R. D. Bhardwaj, S. Bernard, S. Zdunek, F. Barnabe-Heider, S. Walsh, J. Zupicich, K. Alkass, B. A. Buchholz, H. Druid, S. Jovinge and J. Frisen, Science, 2009, 324, 98–102 CrossRef CAS
- K. Jung and R. Reszka, Adv. Drug Delivery Rev., 2001, 49, 87–105 CrossRef CAS
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185–229 CrossRef CAS
- C. E. Myers, W. P. McGuire, R. H. Liss, I. Ifrim, K. Grotzinger and R. C. Young, Science, 1977, 197, 165–167 CAS
- F. M. Yakes and B. Van Houten, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 514–519 CrossRef CAS
- Q. M. Chen, V. C. Tu, Y. Wu and J. J. Bahl, Arch. Biochem. Biophys., 2000, 373, 242–248 CrossRef CAS
- J. H. Doroshow, S. Akman, F. F. Chu and S. Esworthy, Pharmacol. Ther., 1990, 47, 359–370 CrossRef CAS
- K. J. Davies and J. H. Doroshow, J. Biol. Chem., 1986, 261, 3060–3067 CAS
- X. Xu, H. L. Persson and D. R. Richardson, Mol. Pharmacol., 2005, 68, 261–271 CAS
- A. Puppo and B. Halliwell, Biochem. J., 1988, 249, 185–190 CAS
- H. Gruen and H. Gorner, Photochem. Photobiol. Sci., 2008, 7, 1344–1352 CAS
- H. Gorner, Photochem. Photobiol., 2003, 77, 171–179 CrossRef CAS
- K. Tickle and F. Wilkinson, Trans. Faraday Soc., 1965, 61, 1981–1990 RSC
- V. Dibrova, V. Klimenko, R. Nurmukhametov and D. Shigorin, J. Appl. Spectrosc., 1990, 53, 242–247 CrossRef CAS
- A. M. Rahman, S. W. Yusuf and M. S. Ewer, Int. J. Nanomedicine, 2007, 2, 567–583 CAS
- S. Bandak, A. Ramu, Y. Barenholz and A. Gabizon, Pharm. Res., 1999, 16, 841–846 CrossRef CAS
- J. W. Lown and H. H. Chen, Can. J. Chem., 1981, 59, 390–395 CrossRef CAS
- M. J. Wood, W. J. Irwin and D. K. Scott, J. Clin. Pharm. Ther., 1990, 15, 291–300 CrossRef CAS
- J. H. Beijnen, O. A. G. J. van der Houwen and W. J. M. Undergerg, Int. J. Pharm., 1986, 32, 123–131 CrossRef CAS
- J. H. Beijnen, G. Wiese and W. J. M. Undergerg, Pharm. Weekbl., Sci. Ed., 1985, 7, 109–116 CAS
-
F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry – Part A: Structure and Mechanism, Springer-Verlag, New York, 2007, 5th edn Search PubMed
-
E. S. Huyser, Free Radical Chain Reactions, Wiley-Interscience, New York, 1970 Search PubMed
- M. Zhou, Z. Diwu, N. Panchuk-Voloshina and R. P. Haugland, Anal. Biochem., 1997, 253, 162–168 CrossRef CAS
- K. J. Reszka, B. A. Wagner, C. P. Burns and B. E. Britigan, Anal. Biochem., 2005, 342, 327–337 CrossRef CAS
- T. M. Kitson and K. E. Kitson, Biochem. J., 1997, 322, 701–708 CAS
- D. J. Simpson, C. J. Unkefer, T. W. Whaley and B. L. Marrone, J. Org. Chem., 1991, 56, 5391–5396 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of chemicals and methods used. See DOI: 10.1039/c2ra20323e |
| This journal is © The Royal Society of Chemistry 2012 |