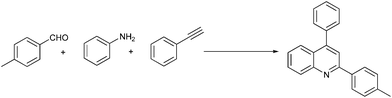
One-pot solvent-free synthesis of quinolines by C–H activation/C–C Bond formation catalyzed by recyclable iron(III) triflate†
Changsheng
Yao
*a,
Bingbin
Qin
b,
Honghong
Zhang
b,
Jun
Lu
b,
Donglin
Wang
b and
Shujiang
Tu
b
aSchool of Chemistry and Engineering, Key Laboratory of Biotechnology for Medicinal Plants, Xuzhou Normal University, Xuzhou, 221116, Jiangsu, P. R. China. E-mail: csyao@xznu.edu.cn.; Fax: 86-516-83500065; Tel: 86-516-83500065
bSchool of Chemistry and Engineering, Jiangsu Key Laboratory of Green Synthetic Chemistry for Functional Materials, Xuzhou Normal University, Xuzhou Jiangsu 221116, P. R. China
First published on 30th January 2012
Abstract
A novel application of highly stable Fe(OTf)3 as an efficient catalyst for carbon–carbon bond formation via the activation of a terminal alkyne C–H bond under solvent-free conditions is described. Notably, this protocol of green synthesis, which produced quinolines from the reaction of amines, aldehydes and terminal aryl alkynes, shows attractive characteristics including concise one-pot conditions, high atom economy, very limited energy consumption, and the sequential catalytic process requires only a catalytic (5 mol%) amount of Fe(OTf)3 with short reaction periods (3 h). Meanwhile, the catalyst was easily recovered from the reaction system and reused smoothly with only a little loss of activity.
Introduction
Recently, applications involving environmentally benign reaction media instead of common organic solvents, many of which are ecologically harmful and require costly remedies, constitute a challenging research area.1 In this sense, an alternative strategy to reduce the waste and adverse impact on the environment is to conduct the reaction under solvent-free conditions.2Quinoline is one of the important heterocyclic compounds and is widely distributed in a vast array of natural products of biological and medical importance, such as antimalarial, antibacterial, antiasthmatic, antihypertensive and anti-inflammatory.3 The best-known methods to introduce a quinoline moiety should be those proposed by Skraup,4 Doebner-von Miller,5 Friedländer,6 and Combes.7 However, the most notable limitations in these procedures include harsh reaction conditions, long reaction times, a large amount of waste, and sometimes the use of refractory substrates such as 2-aminobenzaldehydes. In view of their unique biological and interesting photochemical properties,8 the development of more expeditious and environmentally benign synthetic approaches from easily available precursors is highly warranted.
Over the past decades, transition metal-catalyzed reactions of terminal alkynes have been developed as a versatile tool to construct the unique structural unit from simple precursors in a single procedure. In particular, the activation of a terminal alkyne C–H bond by transition-metal catalysts is often used to facilitate C–N and C–C bond-forming reactions. As a Grignard-type reaction, a recent advance in this field described a highly efficient three-component coupling reaction of aldehydes, alkynes, and amines using several noble transition-metal catalysts and organic solvents. Silver salts,9 a Cu/RuII bimetallic system,10 Ir complexes,11 and copper salts12 have all been used for affording propargyl amines, which are key intermediates in the construction of quinoline derivatives. Furthermore, these two transformations were mediated to synthesize quinolines in a single synthetic operation via a multi-component, AuCl3/CuBr catalytic system.13
However, the high cost and toxic nature of these metal catalysts have somewhat placed limitations on large-scale applications. In this respect, the development of inexpensive, easily available and nontoxic catalysts is highly attractive for chemical synthesis. Much attention has been paid to iron as a promising alternative catalyst for different organic reactions due to its high abundance, low price, non-toxicity, and environmentally well-suited characters. A variety of iron-catalyzed oxidations,14 hydrogenations,15 rearrangements,16 and epoxidations17 have been disclosed and developed successfully. Recently, a FeCl3-catalyzed synthesis of quinolines in toluene was reported in good yields (56–95%) using long reaction times (24 h).18 It's worth noting that the iron-catalyzed methodology is a very efficient process that is now able to compete with the precious metal-catalyzed one. In contrast, iron(III) triflate is a better potential Lewis acid catalyst, which is highly stable in water and does not decompose under aqueous conditions. The non-hygroscopic nature, high catalytic activity, short reaction periods, facile separation and reuse of this catalyst are other advantages over other conventional Lewis acids. Actually, we did find unprecedented reactivity in the cationic iron catalyst Fe(OTf)3, described by the groups of Takaki.19
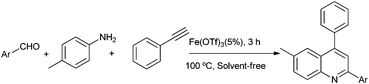
To continue our work on the multi-component synthesis of heterocyclic compound library with high diversity,20 herein we wish to disclose an eco-friendly and efficient solvent-free system for the green synthesis of substituted quinolines based on a novel cationic iron catalyst Fe(OTf)3via a three-component domino reaction of aldehydes, amines and terminal alkynes (Scheme 1). The sequential catalytic process requires only 5% of the amount of catalyst at the same time to shorten the reaction time (3 h) and the activity of easily recoverable catalyst Fe(OTf)3 was still maintained at a desirable level.
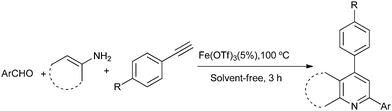 | ||
| Scheme 1 Synthesis of quinolines via a three-component reaction. | ||
Results and discussion
We initialized the catalytic activity of Fe(OTf)3 using aniline, 4-methyl benzaldehyde with phenylacetylene as the model substrates in toluene under an air atmosphere at 110 °C for 24 h, and the result is summarized in Table 1. To our delight, the three-component coupling reaction, in the presence of 10 mol% of Fe(OTf)3, proceeded smoothly and generated bioactive quinoline in 75% yield (Table 1, entry 2). By contrast, switching to FeCl3 as the catalyst also resulted in the desired product in 72% yield (Table 1, entry 1).18 As expected, the catalytic effect of metal cations is even more pronounced for this reaction. Unfortunately, the quinoline product was obtained only in 47% yield when the reaction was catalyzed by Fe(OTf)2 (Table 1, entry 3). Additionally, the results show that other iron salts, based on metal salts with non-nucleophilic anions, displayed poor activity to this reaction under the same conditions (Table 1, entries 4–6).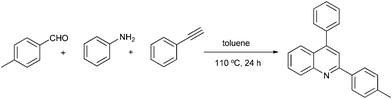
| Entry | Catalyst (10%) | Yield (%)b |
|---|---|---|
| a 1.0 mmol 4-methyl benzaldehyde, 1.05 mmol aniline and 1.5 mmol phenylacetylene. b Isolated yield calculated on the basis of aldehyde. | ||
| 1 | FeCl3 | 72 |
| 2 | Fe(OTf)3 | 75 |
| 3 | Fe(OTf)2 | 47 |
| 4 | FeSO4 | 29 |
| 5 | Fe(NO3)3 | Trace |
| 6 | Fe2(SO4)3 | Trace |
Inspired by the result, we next screened the effect of solvent on the reaction by using 10 mol% of Fe(OTf)3 as catalyst (Table 2, entries 1–8, and 11). The choice of solvent did influence the activity of the catalytic system: the solvent-free system (Table 2, entry 11) exhibits better yield and greater reaction rate than other counterparts, given the desired product in 77% yield at 110 °C for 2 h. Some classical solvents such as toluene, benzene, 1,2-dichloroethane (DCE), ethanol, acetonitrile and tetrahydrofuran (THF) were inferior and generated quinoline in 75, 66, 39, 34, 36 and 38% yields for 24 h, respectively (Table 2, entries 1–6). Unfortunately, no product formation was observed when the reaction was carried out in dimethyl sulfoxide (DMSO), or N,N-dimethylformamide (DMF) (Table 2, entries 7–8).
| Entry | Solvent/T/°C | Time/h | Yieldb (%) |
|---|---|---|---|
| a 1.0 mmol 4-methyl benzaldehyde, 1.05 mmol aniline and 1.5 mmol phenylacetylene. b Isolated yield calculated on the basis of aldehyde. c 5% mol Fe(OTf)3 was used. d 2% mol Fe(OTf)3 was used. | |||
| 1 | Toluene/reflux | 24 | 75 |
| 2 | Benzene/reflux | 24 | 66 |
| 3 | DCE/reflux | 24 | 39 |
| 4 | C2H5OH/reflux | 24 | 34 |
| 5 | CH3CN/reflux | 24 | 36 |
| 6 | THF/reflux | 24 | 38 |
| 7 | DMSO/110 | 24 | 0 |
| 8 | DMF/110 | 24 | 0 |
| 9 | solvent-free/90 | 4 | 70 |
| 10 | solvent-free/100 | 2.5 | 82 |
| 11 | solvent-free/110 | 2 | 77 |
| 12c | solvent-free/100 | 3 | 84 |
| 13d | solvent-free/100 | 4 | 75 |
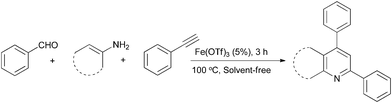
To further screen for the practical temperature of the synthesis, the previous reaction was carried out under solvent-free conditions at 90 and 100 °C. It was very surprising that the solvent-free reaction at 100 °C proceeded in excellent yield (82%, Table 2, entry 10), but the yield decreased when the reaction was carried out at 90 °C (70%, Table 2, entry 9). Meanwhile, the amount of catalyst was also investigated. It is gratifying that we have succeeded in reducing the catalyst loading to 5 mol% (relative to the 4-methyl benzaldehyde) without adversely affecting the product yield (84%, Table 2, entry 12). However, employment of 2 mol% of the catalyst provided the desired product in lower yield (75%, Table 2, entry 13).
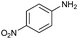
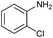
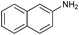
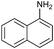
Encouraged by the efficiency of the methodology described above, the scope and generality of the reaction was examined with Fe(OTf)3 (5 mol%) under solvent-free conditions at 100 °C. Firstly, a broad variety of substituted amines were investigated to be reacted with benzaldehyde and phenylacetylene. The results are listed in Table 3. To our delight, we found this transformation to be very general for substituted amines. Electron-rich and electron-poor groups attached to the benzene ring were uneventful, and furnished the desired quinolines in excellent yields (Table 3). Also, the procedure can be extended to α- and β-naphthylamine, although the products were obtained in moderate yields (Table 3, entries 7–8).
| Entry | Amines | Yieldb (%) |
|---|---|---|
| a Reaction conditions: benzaldehyde (1.0 mmol), amine (1.05 mmol), phenylacetylene (1.5 mmol), Fe(OTf)3 (0.05 mmol), solvent-free, 100 °C, under an air atmosphere. b Isolated yield calculated on the basis of aldehyde. | ||
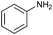
| 1 |
|
88 |
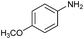
| 2 |
|
78 |
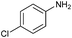
| 3 |
|
82 |
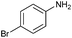
| 4 |
|
81 |
| 5 |
|
69 |
| 6 |
|
71 |
| 7 |
|
82 |
| 8 |
|
75 |
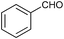
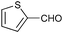
To further probe the scope of the procedure, various aldehydes were surveyed under the same reaction conditions. As shown in Table 4, most aldehydes displayed good activities in this reaction. Similar to electron-rich phenyl ring (entries 6 and 7), substrates bearing halogen, 4-Br, 4-Cl, and 4-F, were also efficiently converted into quinolines in 77%, 79%, and 84% yields, respectively (entries 2–4). And electron-poor arene possessing two chlorine atoms at position 3 and 4 took part in the reaction, giving the desired product in high yield (82%, entry 5). In addition, the thiophene-2-carbaldehyde was also subjected to the reaction conditions, and the corresponding product was obtained in moderate to excellent yield (Table 4, entry 8).
| Entry | Aryl aldehydes | Yieldb (%) |
|---|---|---|
| a Reaction conditions: aldehydes (1.0 mmol), p-toluidine (1.05 mmol), phenylacetylene (1.5 mmol), Fe(OTf)3 (0.05 mmol), solvent-free, 100 °C, under an air atmosphere. b Isolated yield calculated on the basis of aldehyde. | ||
| 1 |
|
80 |
| 2 |
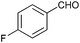
|
84 |
| 3 |
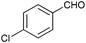
|
79 |
| 4 |
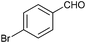
|
77 |
| 5 |
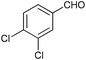
|
82 |
| 6 |
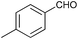
|
86 |
| 7 |
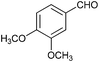
|
88 |
| 8 |
|
85 |
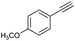
Subsequently, the selectivity of this chemistry was further examined using substrates with substituted anilines, benzaldehydes and terminal phenylacetylenes. As shown by the results in Table 5, this procedure worked delightfully well, and gave a range of products in good yields. It was apparent that p-methoxyphenylacetylene was less reactive and provided quinolines in moderate yields, compared with phenylacetylene and p-fluorophenylacetylene. Possibly, electronic factors affected the selectivity, and the fluorine substituent at the phenyl ring 4-carbon expectedly facilitated the selective formation of a ring. However, the methoxyl group at the ortho-position of acetenyl group deactivated the alkyne.
| Entry | Amines | Aryl aldehydes | Alkynes | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: aldehydes (1.0 mmol), amines (1.05 mmol), alkynes (1.5 mmol), Fe(OTf)3 (0.05 mmol), solvent-free, 100 °C, under an air atmosphere. b Isolated yield calculated on the basis of aldehyde. | ||||
| 1 |
|
|
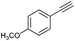
|
72 |
| 2 |
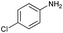
|
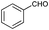
|
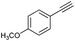
|
70 |
| 3 |
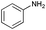
|
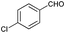
|

|
66 |
| 4 |
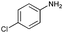
|
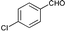
|
|
71 |
| 5 |
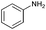
|
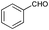
|
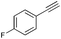
|
81 |
| 6 |
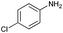
|
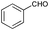
|
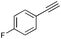
|
76 |
| 7 |
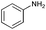
|
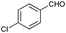
|
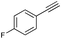
|
78 |
| 8 |
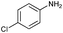
|
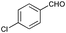
|
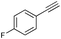
|
80 |
| 9 |
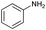
|
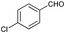
|
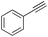
|
71 |
| 10 |
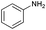
|
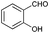
|
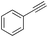
|
87 |
| 11 |
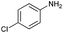
|
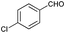
|
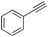
|
75 |
An important feature of the cationic iron catalyst is the easy and reliable separation based on its stability in water and non-decomposing under aqueous conditions. In this study, Table 6 shows the recycling experiments of this catalysis upon the reaction of aniline, 4-methyl benzaldehyde and phenylacetylene.21 After completion, the reaction mixture was completely dissolved in dichloromethane, and then washed with cold water (3 × 10 mL). The catalyst was recovered from the aqueous layer via evaporation of the water, and the recovered catalyst was reused for at least three cycles in subsequent reactions.
| Runb | Recovery rate of catalyst (%) | Yield (%) |
|---|---|---|
| a 1.0 mmol 4-methyl benzaldehyde, 1.05 mmol aniline and 1.5 mmol phenylacetylene. b Reaction conditions: solvent-free, 100 °C, under an air atmosphere. | ||
| 1 | 92 | 80 |
| 2 | 91 | 78 |
| 3 | 92 | 75 |
| 4 | 92 | 74 |
Together with several literature publications,18 a possible mechanism of the iron-catalyzed synthesis of quinolines from three-component coupling of aldehyde, alkyne, and amine is illustrated in Scheme 2. Generally, the first proposed step is the formation of intermediate A, which is generated by successive complexation of imine and alkyne to Fe(III). In this process, imine is attacked by activated phenylacetylene to give the propargylamine B, and then the nucleophilic attack of the ortho-position of the aniline on the iron acetylide gives cyclic intermediate C, which undergoes oxidative aromatization by O2 in air to give the final product. Because the propargylic amine was not detected in the sequential catalytic process, we consider that the intermediate B experiences a cooperative process with the subsequent hydroarylation reaction.
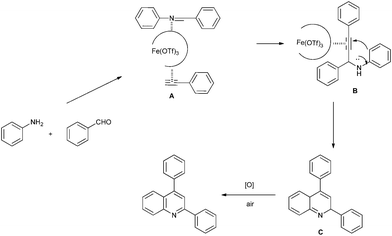 | ||
| Scheme 2 A proposed mechanism for the Fe(OTf)3-catalyzed synthesis of 2,4-diphenylquinoline from aniline, benzaldehyde and phenylacetylene. | ||
To verify the structure of the products, Table 4, entry 5 and Table 5, entry 10 were selected as representative compounds and characterized by X-ray crystallography as shown in Fig. 1 and Fig. 2.22,23
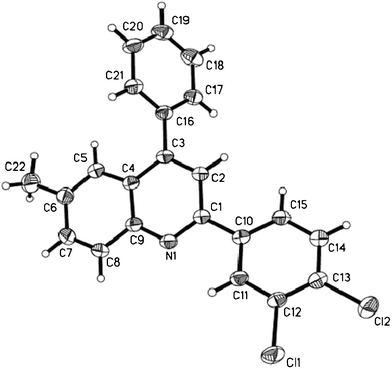 | ||
| Fig. 1 ORTEP diagram of Table 4, entry 5. | ||
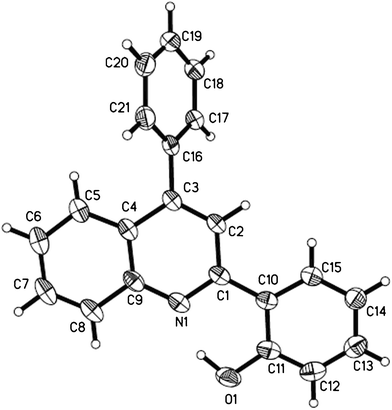 | ||
| Fig. 2 ORTEP diagram of Table 5, entry 10. | ||
Conclusions
To summarize, an environmentally friendly, economical and efficient method was successfully developed for synthesizing quinoline derivatives using a novel Lewis acid and easily recoverable catalyst Fe(OTf)3 under solvent-free conditions via one-pot three-component coupling of aldehydes, alkynes, and amines. The combination of the cationic iron catalyst, solvent-free system and a multicomponent reaction approach provided the products in excellent selectivities and yields in short reaction times (3 h). Of special importance, the inexpensive and non-hygroscopic iron complex Fe(OTf)3 has been proven to be a new effective catalyst, or superior to the triflates of precious metals, such as silver and copper, or triflic acid alone for activating a terminal alkyne C–H bond even at low catalyst loadings (5 mol%).16 Further investigations on the application of this kind of catalyst are underway in our laboratory.Experimental section
Catalyst preparation24
Iron(III) triflate was prepared in the following way: triflic acid (12.3 g, 81.6 mmol) was gradually added to crystals of Fe(NO3)3·9H2O (10.0 g, 24.8 mmol), and the mixture was heated to 250 °C in a porcelain vessel placed on a sand bath until the evolution of nitrogen oxides and water was not noticed. The white crystalline material (12.3 g, 99% yield) was allowed to cool in a desiccator, and used without further purification.General procedure for the iron-catalyzed three-component coupling reactions of aldehydes, alkynes, and amines
To a 5 mL flask was sequentially added Fe(OTf)3 (25.1 mg, 0.05 mmol), aryl aldehyde (1.0 mmol), aryl amine (1.05 mmol), and aryl acetylene (1.5 mmol) under an air atmosphere. The reaction vessel was placed in an oil bath at 100 °C, and then the mixture was stirred until the substrates had been consumed completely. The reaction system was cooled to room temperature and diluted with dichloromethane, and then washed with cold water (3 × 10 mL). The organic phase was separated, and the aqueous layer was washed with dichloromethane (3 × 10 mL). Concentration of the combined organic layer afforded the crude product, which was further purified by flash chromatography on silica gel (eluent: petroleum ether/AcOEt = 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). The catalyst was recovered from the aqueous layer via evaporation of the water, and dried at 70 °C for 2 h to give pure Fe(OTf)3 in 92% recovery. The recovered catalyst was reused for the next run in the same way. The identity and purity of products was confirmed by 1H and 13C NMR spectroscopy. See the ESI† for full details.
:1, v/v). The catalyst was recovered from the aqueous layer via evaporation of the water, and dried at 70 °C for 2 h to give pure Fe(OTf)3 in 92% recovery. The recovered catalyst was reused for the next run in the same way. The identity and purity of products was confirmed by 1H and 13C NMR spectroscopy. See the ESI† for full details.
Acknowledgements
We are grateful for financial support by A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, the Major Basic Research Project of the Natural Science Foundation of the Jiangsu Higher Education Institutions (09KJA430003), Natural Science Foundation of Xuzhou City (XM09B016), Graduate Foundation of Xuzhou Normal University (2010YLB029) and Qing Lan Project (08QLT001).References
-
(a)
J. Clark, D. Macquarrie, Handbook of Green Chemistry & Technology, Blackwell Science Ltd., Oxford, UK, 2002, pp. 540 Search PubMed
; (b) K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025 CrossRef CAS
-
(a)
K. Tanaka, F. Toda, Solvent-Free Organic Synthesis, John Wiley & Sons, Chichester, UK, 2003, pp. 300 Search PubMed
-
(a) O. Billker, V. Lindo, M. Panico, A. E. Etienne, T. Paxton, A. Dell, M. Rogers, R. E. Sinden and H. R. Morris, Nature, 1998, 392, 289 CrossRef
-
(a) Z. H. Skraup, Ber., 1880, 13, 2086
-
(a) A. R. Mackenzie, C. J. Moody and C. W. Rees, Tetrahedron, 1986, 42, 3259 CrossRef CAS
-
(a) P. Friedläender and C. F. Gohring, Ber. Dtsch. Chem. Ges., 1883, 16, 1833 CrossRef
-
(a) W. S. Johnson and F. J. Mathews, J. Am. Chem. Soc., 1944, 66, 210 CrossRef CAS
- J. I. Kim, I.-S. Shin, H. Kim and J.-K. Lee, J. Am. Chem. Soc., 2005, 127, 1614 CrossRef CAS
- C.-M. Wei, Z.-G. Li and C.-J. Li, Org. Lett., 2003, 5, 4473 CrossRef CAS
- C.-J. Li and C.-M. Wei, Chem. Commun., 2002, 268 RSC
-
(a) S. Sakaguchi, T. Kubo and Y. Ishii, Angew. Chem., Int. Ed., 2001, 40, 2534 CrossRef CAS
-
(a) L. Shi, Y.-Q. Tu, M. Wang, F.-M. Zhang and C.-A. Fan, Org. Lett., 2004, 6, 1001 CrossRef CAS
- F. Xiao, Y. Chen, Y. Liu and J. Wang, Tetrahedron, 2008, 64, 2755 CrossRef CAS
-
(a) M. Nakanishi and C. Bolm, Adv. Synth. Catal., 2007, 349, 861 CrossRef CAS
-
(a) S. C. Bart, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 13794 CrossRef
- G. Zhang, Q. Liu, L. Shi and J. Wang, Tetrahedron, 2008, 64, 339 CrossRef CAS
-
(a) F. G. Gelalcha, B. Bitterlich, G. Anilkumar, M. K. Tse and M. Beller, Angew. Chem., Int. Ed., 2007, 46, 7293 CrossRef CAS
- K. Cao, F.-M. Zhang, Y.-Q. Tu, X.-T. Zhuo and C.-A. Fan, Chem.–Eur. J., 2009, 15, 6332 CrossRef CAS
- K. Komeyama, R. Igawa and K. Takaki, Chem. Commun., 2010, 46, 1748 RSC
-
(a) B. Jiang, S.-J. Tu, P. Kaur, W. Wever and G. Li, J. Am. Chem. Soc., 2009, 131, 11660 CrossRef CAS
-
(a) Y.-Y. Zhang, J.-D. Lin, X.-L. Xu and J.-H. Li, Synth. Commun., 2010, 40, 2556 CrossRef CAS
- The single-crystal growth was carried out in ethanol at room temperature. X-ray crystallographic analysis was performed using a Rigaku Saturn diffractometer. Crystal data for Table 4, entry 5: C22H15Cl2N, M = 364.25, monoclinic, a = 8.486(3) Å, b = 15.397(5) Å, c = 14.367(4) Å, α = 90.00°, β = 111.573(15)°, γ = 90.00°, V = 1745.7(10) Å3, T = 293(2)K, space group P21/c, Z = 4, 12183 reflections measured, 4668 independent reflections (Rint = 0.0341). The final R1 values were 0.0461 (I > 2σ(I)). The final wR(F2) values were 0.1347 (I > 2σ(I)). The final R1 values were 0.0686 (all data). The final wR(F2) values were 0.1610 (all data). CCDC 842657
contains the supplementary crystallographic data of this compound. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- The single-crystal growth was carried out in ethanol at room temperature. X-ray crystallographic analysis was performed using a Rigaku Saturn diffractometer. Crystal data for Table 5, entry 10: C21H15NO, M = 297.34, a = 13.740(2) Å, b = 7.0021(12) Å, c = 15.550(3) Å, α = 90.00°, β = 95.820(4)°, γ = 90.00°, V = 1488.3(4) Å3, T = 293(2)K, Z = 4 , 7918 reflections measured, 2612 independent reflections (Rint = 0.0312). R1 = 0.0416 (I > 2σ(I)). The final wR(F2) values were 0.1177 (I > 2σ(I)). The final R1 values were 0.0599 (all data). The final wR(F2) values were 0.1271 (all data). CCDC 842682 contains the supplementary crystallographic data of this compound. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- S. Ichikawa, I. Tomita, A. Hosaka and T. Sato, Bull. Chem. Soc. Jpn., 1988, 61, 513 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC reference numbers 842657, 842682. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra20172k |
| This journal is © The Royal Society of Chemistry 2012 |