Nanoscale assembly, morphology and screening effects in nanorods of newly synthesized substituted pentacenes
Sabine-Antonia
Savu
a,
Maria Benedetta
Casu
*a,
Simon
Schundelmeier
a,
Sabine
Abb
a,
Christina
Tönshoff
b,
Holger F.
Bettinger
b and
Thomas
Chassé
a
aInstitute of Physical and Theoretical Chemistry, University of Tuebingen, Auf der Morgenstelle 18 D-72076, Tuebingen, Germany. Fax: +49 7071 29 5490; Tel: +49 7071 29 76252 Web: http://www.condensed-matter.uni-tuebingen.de
bInstitute of Organic Chemistry, University of Tuebingen, Auf der Morgenstelle 18, D-72076, Tuebingen, Germany
First published on 13th April 2012
Abstract
We report our investigation on the nanorods of two newly synthesized substituted pentacenes, δ4-substituted (2,3-X2-9,10-Y2) pentacene with X = Y = methoxy group (MOP) and X = F, Y = methoxy (MOPF), by using X-ray photoemission spectroscopy (XPS), near edge X-ray absorption fine structure (NEXAFS), and atomic force microscopy (AFM). The nanorods were deposited on Au(111) single crystals. Energy dependent photoemission spectra show complex features, including a rich satellite structure that we have analyzed in detail by using a best-fit procedure applying constraints based on stoichiometry, electronegativity, and bond strength. This analysis reveals the presence of surface core level shifts due to the high electronegativity of the fluorine atoms. The distinctive features of growth and morphology of the nanorods are subjected to a template effect by the substrate lattice geometry, leading to morphological well-organized assemblies. Fluorine atoms play an important role not only in the electronic structure but also in the morphology of the nanorod assemblies.
Introduction
Organic materials were considered very appealing in the past, not only because of a pure academic interest, but also because of their promising characteristics towards electronic applications.1 These materials have kept their promise and they have reached the market in a very short period of time, since the first organic light emitting device was demonstrated.2 Their properties are very attractive in view of their use in a large number of applications where low costs, chemical flexibility and energy saving technologies play the major roles and organic molecules may achieve a different or better performance than inorganic semiconductors.3 Pentacene is one of the most widely used organic active media due to its physical, chemical and morphological properties. Its high charge carrier mobility and the possibility to grow highly oriented thin films lead to a strong improvement in device performances.4,5 The tuning of its electronic structure, HOMO (highest occupied molecular orbital)–LUMO (lowest unoccupied molecular orbital) gap, optical and transport properties by using appropriate substituents is a feasible way to broaden its use towards new applications:6,7 the compatible molecular structure of pentacene and its substituted versions makes possible a suitable structural coupling in view of the formation of p–n junctions, as seen with perfluoropentacene.8 Substituted pentacenes are also very popular as molecular walkers, i.e., molecules that are capable of moving unidirectionally across a substrate violating its symmetry,9 opening the way to using them, for example, as molecular cargo. This property can be tuned with different methods and specifically with substitution, also on the ring.9Here, we focus our investigation on two newly synthesized substituted pentacenes: δ4-substituted (2,3-X2-9,10-Y2) pentacene with X = Y = methoxy group (MOP) and X = F, Y = methoxy (MOPF)7 reporting the results obtained by using X-ray photoemission spectroscopy (XPS), near edge X-ray absorption fine structure (NEXAFS), and atomic force microscopy (AFM) on MOP and MOPF molecules deposited on Au(111).
We analyze the rich satellite structure in the energy dependent photoemission spectra and also we find evidence for surface core level shifts due to the high electronegativity of the fluorine atoms. In addition, the deposited systems show distinctive nanorod morphology (see inset in Fig. 1) whose growth is influenced by the substrate geometry. The dimensionality of the nanorods adds a further source of interest to these molecules besides their electronic structure. The self assembly of ordered structures is an important issue in nanotechnology, together with low dimensionality in morphology this can lead to important applications in devices, as seen for thiophene-phenylene cooligomers10 or toward inter-digitated heterostructures.
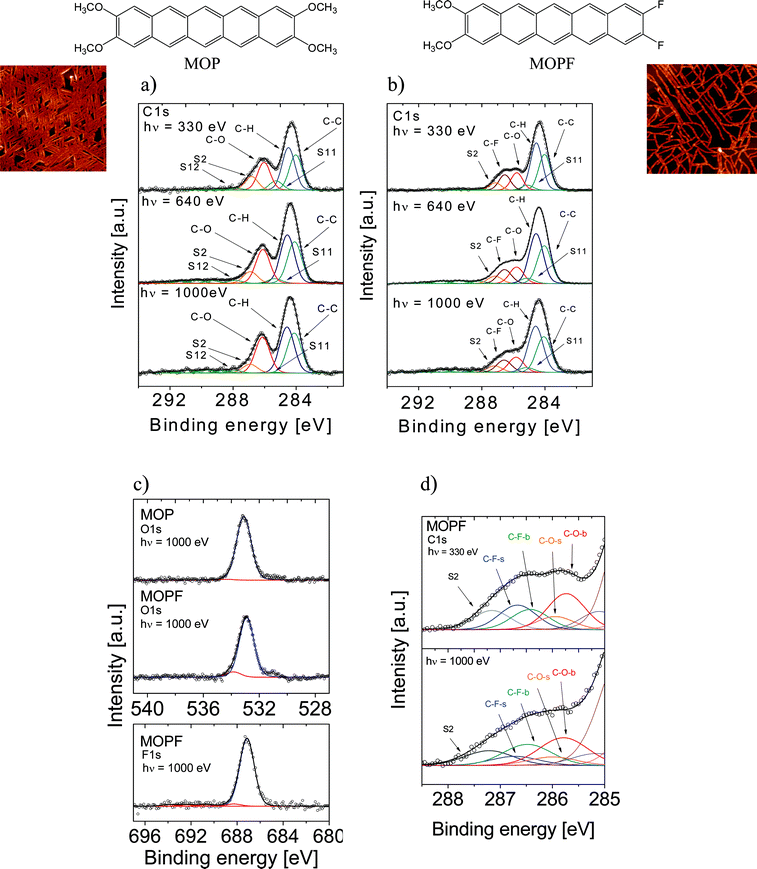 | ||
| Fig. 1 Photon energy dependent core level photoemission spectra together with their relative fits and chemical structures of the molecules for MOP (nominal thickness: 160 Å) and MOPF (nominal thickness: 52 Å). Three different excitation energies were used, as indicated. (a) MOP C1s core level spectra. (b) MOPF C1s core level spectra. (c) MOP and MOPF O1s core level spectra (upper panel) and MOPF F1s core level spectra (lower panel). (d) SCLS fit components (b = bulk, s = surface). In the insets two typical 5 μm × 5 μm AFM images of the nanorods are shown. | ||
Experimental section
The photoelectron and absorption spectra were recorded at the UE52-PGM undulator beamline at BESSY (Berlin). This beamline is characterized by a plane grating monochromator. The main chamber (base pressure 4 × 10−10 mbar) is equipped with a standard twin anode X-ray source, and a SCIENTA R4000 electron energy analyzer. The measurements were carried out in a single bunch (ring current at injection = 20 mA, cff = 2, 5, 20 μm exit slit, analyzer resolution = 0.1 eV). The C1s photoelectron spectra were taken with photon energies of 330, 640, and 1000 eV. NEXAFS spectra were normalized by taking the ring current and the clean substrate signal into account. Afterwards all spectra were scaled to give an equal edge jump.The nanorod preparation occurred in situ under ultra high vacuum (UHV) conditions by organic molecular beam deposition. The substituted pentacene molecules were evaporated on a Au(111) single crystal surface, which was cleaned with several cycles of sputtering and annealing. As a standard procedure, the cleaning cycles were repeated until XPS showed no trace of contaminants, ultraviolet photoelectron spectroscopy (UPS) the appropriate work function, and low energy electron diffraction (LEED) the expected pattern. No deposition has been performed on contaminated substrates. The substrate was kept at room temperature. The deposition rate was 1 and 0.7 Å min−1 for MOP and MOPF, respectively. It was determined by using a quartz microbalance and cross-checked by using the attenuation of the XPS substrate signal after deposition. Atomic force microscope studies were performed ex-situ with a Digital Instruments Nanoscope III Multimode AFM. The experiments were carried out in tapping mode. No degradation of the samples was observed on the time scale of all the presented experiments.
Results and discussion
Fig. 1 shows the excitation energy dependent photoemission core level spectra of MOP and MOPF. The C1s experimental curves of both molecules show a main line at 284.3 eV. MOP spectra show a further peak at 286.1 eV. In MOPF curves a broad feature is visible at higher binding energies. Basing our assignment on a pure electronegativity argument, the main spectral feature contains contributions from photoelectrons coming from the ring carbon atoms without any bonding to heteroatoms. The structures at higher binding energies are due to the signals from carbon atoms, which are bonded to oxygen and/or fluorine. The high electronegative bonding partners withdraw electrons and thus reduce the charge density that results in an increased binding energy.Performing a curve fit (using Unifit 2010)11 and looking very carefully at its details allows a deeper insight into the photoemission spectra. The C1s core level photoemission lines were fitted with Voigt profiles (constant Lorentzian width of 0.08 eV).12,13 The binding energies of the peak components were kept constant for all excitation energies (see Table 1 and 2). Because of stoichiometric boundary conditions, the intensity ratio of the C1s main line and the C–O line plus the corresponding satellites should be 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 for MOP; while for MOPF the intensity ratio of the C1s main line, the C–O line, and the C–F line plus the corresponding satellites should be 18:4:2. As a result of the D2h symmetry of MOP, seven different C1s core level features could be expected: one for each non equivalent carbon species. Analogously, twelve different carbon features are expected for MOPF (C2v symmetry). Within our experimental resolution, identifying all of them by fitting would be quite speculative. Thus, we have limited the boundary fitting conditions by taking into account three different carbon atom sites in MOP, and four in MOPF: (i) carbon atoms which are bonded to heteroatoms (C–F, C–O), (ii) carbon atoms bonded to carbon and hydrogen atoms (C–H), and (iii) carbon atoms of the inner rings which are only bonded to carbon atoms (C–C) (see molecular structures in Fig. 1). As a consequence of our procedure, we can identify two contributions in the main peak: namely due to signals from the C–C-species (lower binding energy, Fig. 1a) and the C–H-species (higher binding energy, Fig. 1a). These two contributions are caused by the different local chemical environments seen by the carbon atoms bound to hydrogen.13–18 We assign the lowest binding energy to the C–C-species as a result of our best fit procedure taking into account the stoichiometry constraints. This assignment indicates a more efficient screening of the core–hole in the internal ring C–C bonds and it may be attributed to the cooperation in the screening of two adjacent rings, i.e., due to a better charge delocalization along this channel.
:8 for MOP; while for MOPF the intensity ratio of the C1s main line, the C–O line, and the C–F line plus the corresponding satellites should be 18:4:2. As a result of the D2h symmetry of MOP, seven different C1s core level features could be expected: one for each non equivalent carbon species. Analogously, twelve different carbon features are expected for MOPF (C2v symmetry). Within our experimental resolution, identifying all of them by fitting would be quite speculative. Thus, we have limited the boundary fitting conditions by taking into account three different carbon atom sites in MOP, and four in MOPF: (i) carbon atoms which are bonded to heteroatoms (C–F, C–O), (ii) carbon atoms bonded to carbon and hydrogen atoms (C–H), and (iii) carbon atoms of the inner rings which are only bonded to carbon atoms (C–C) (see molecular structures in Fig. 1). As a consequence of our procedure, we can identify two contributions in the main peak: namely due to signals from the C–C-species (lower binding energy, Fig. 1a) and the C–H-species (higher binding energy, Fig. 1a). These two contributions are caused by the different local chemical environments seen by the carbon atoms bound to hydrogen.13–18 We assign the lowest binding energy to the C–C-species as a result of our best fit procedure taking into account the stoichiometry constraints. This assignment indicates a more efficient screening of the core–hole in the internal ring C–C bonds and it may be attributed to the cooperation in the screening of two adjacent rings, i.e., due to a better charge delocalization along this channel.
| EB/eV (hν = 1000) | Intensity/% | EB/eV (hν = 640) | Intensity/% | EB/eV (hν = 330) | Intensity/% | |
|---|---|---|---|---|---|---|
| C–C | 284.12 | 27.84 | 284.10 | 26.99 | 284.03 | 26.44 |
| C–H | 284.58 | 32.05 | 284.56 | 31.43 | 284.49 | 31.86 |
| S11 | 285.37 | 0.56 | 285.35 | 3.03 | 285.28 | 7.04 |
| C–O | 286.13 | 24.97 | 286.11 | 21.94 | 286.04 | 20.78 |
| S2 | 286.92 | 6.19 | 286.90 | 7.6 | 286.83 | 10.24 |
| EB/eV (hν = 1000) | Intensity/% | EB/eV (hν = 640) | Intensity/% | EB/eV (hν = 330) | Intensity/% | |
|---|---|---|---|---|---|---|
| C–C | 284.07 | 27.23 | 284.05 | 26.96 | 284.03 | 27.34 |
| C–H | 284.58 | 35.44 | 284.56 | 35.63 | 284.54 | 36.13 |
| S11 | 285.16 | 3.71 | 285.13 | 3.79 | 285.12 | 3.83 |
| C–O | 285.82 | 11.68 | 285.80 | 11.77 | 285.78 | 13.36 |
| C–F | 286.58 | 9.40 | 286.56 | 9.94 | 286.54 | 11.77 |
| S2 | 287.19 | 4.95 | 287.17 | 5.28 | 287.15 | 5.67 |
We note here that our assignment perfectly agrees with ref. 13. In some previous work,14–17 the assignment is opposite (i.e., the lower contributions are assigned to C–H-species) for pentacene and perylene-tetracarboxylic acid-dianhydride (PTCDA). This assignment requires a fit with a larger number of contributions, for example in pentacene the single main peak has been fitted with four contributions.14 The discrepancy between the two cases has never been clearly discussed even in cases of crossed references.16,17 Thus, we point out that our fit procedure, due to the presence of the substituents, has more stringent stoichiometric requirements that must be satisfied making the assignment reliable. In addition, our assignment agrees with the XPS C1s spectra obtained for hydrogenated single wall carbon nanotubes where the C–H bond formation leads to a new feature in the higher binding energy range.18
The effect of the electronegative heteroatoms, oxygen and fluorine, is mirrored by the fact that the binding energy of the four external carbon atoms belonging to the terminal rings (left and right) of the pentacene backbone is higher than the binding energy of the remaining atoms of the molecular backbone. Thus, oxygen and fluorine atoms do not only affect the electron density of their neighbouring atoms, they also indirectly affect the electron density in the end rings.
Both molecules show a widely spread satellite structure, which is typical for acenes.19 As a result of the core–hole formation the symmetry is reduced and a larger number of non-equivalent carbon atoms should be considered. The ionization at different carbon sites may give different contributions to the shake-up spectra. The S11 satellite can be related to the first HOMO–LUMO shake up.19 Its energy position with respect to the main line (1.3 eV for MOP and 1.1 eV for MOPF) is lower than the optical gap (2.31 eV for MOP and 2.19 eV for MOPF).7 This is a typical effect in the HOMO–LUMO shake up satellites of polyacenes,19 and it is also seen in perylene-based molecules.20–22 Satellite intensities in polyacenes have been discussed in full detail by Rocco et al.19 The reduced binding energy with respect to the optical gap of the first HOMO–LUMO shake up satellite in large acenes is caused by the enhanced screening of the core–hole due to the delocalization of the aromatic system. Rocco et al. also point out that the total wave functions of the 1s hole states (ionic ground states) in large acenes resemble the neutral ground state wave functions in polyacenes less than in benzene. Thus, the overlap integrals on which the intensity of the main line depends, obeying the monopole selection rule, decrease and the intensity of the satellites must increase with respect to the main line when keeping the C1s cross section constant.19
A change in photon energy as performed in the present experiment implies a change in the C1s cross section, increasing the complexity of the screening effects, as reflected by our experiment and our fit procedure. We note in Fig. 1a and 1b that the S11 intensity decreases with an increasing photon energy while the intensities of the remaining satellites show the opposite behaviour (the spectra have been normalized so that the main C1s lines have the same intensity to facilitate a comparison). This gives a hint about the fact that the S11 intensity is related to the dipole excitation of a core electron to the LUMO accompanied by the monopole ionization of the valence electron: this shake up contribution is near to the ionization threshold region and decreases with the increase in energy,23,24 as observed in our spectra.
S2 is the satellite related to the C–O species. These satellites are very intense, but this is not surprising. Satellite intensities also depend on donor–acceptor interactions.13 In the MOP molecule the methoxy groups act as strong electron donors and this determines the high S2 intensity. MOPF represents a so-called push pull system:7 on one hand the methoxy group acts again as an electron donor, and on the other hand fluorine acts as an electron acceptor. Consequently, S2 is again very intense.
The satellites at the higher binding energy can be assigned to the ring carbon atoms without bonding to oxygen and fluorine. These assignments are in agreement with the stoichiometric ratio and with previous analyses of pentacene XPS spectra.19
Apart from all effects so far analyzed, a small difference in the curve shape depending on the excitation energy is recognizable in MOP. In analogous experiments performed on PTCDA, coronene, and metal free phthalocyanine (H2Pc), no change in shape was observed.25,26 These organic molecules represent systems with different degrees of intermolecular interaction ranging from a stronger one like in PTCDA or H2Pc and to a certainly weaker interacting system like coronene. It is worth noting that the H2Pc thin film showed tilted molecules with respect to the substrate, while spectra of PTCDA, a perylene-based molecule that also contains oxygen atoms, were measured in thin films with perfectly flat lying molecules with respect to the substrate. The results on PTCDA and H2Pc demonstrate that electronegativity alone cannot account for such an effect in MOP. In order to explore the reason leading to its different behaviour, we have measured NEXAFS spectroscopy at the C K edge to gain structural information on the nanorod assembly (Fig. 2). NEXAFS spectra can be quantitatively analyzed to determine the orientation of the molecular plane with respect to the substrate, because of the polarization dependence that stems from the dipole selection rules.27 The calculated molecular orientation with respect to the substrate is 43° and 38° for MOP and MOPF, respectively. The NEXAFS signal is integrated over the area sampled by the incident spot and the obtained molecular orientation is an average value. However, the present spectra are very similar to the spectra obtained for pentacene on Au(111).28,29 This similarity helps in their interpretation: on Au(111) pentacene molecules optimize their free energy adopting a recumbent position.28,29 In particular Cantrell et al.29 have summarized the information on pentacene structure reported in more than 50 papers to establish a correlation between preparation conditions and tilt angle. Their tables show that the recumbent position is most likely to occur on Au(111). Our NEXAFS results can be interpreted with MOP and MOPF molecules adopting a recumbent position in the nanorods (i.e., molecules lying with their long axis parallel to the substrate and with a certain tilt angle of the short axis) as seen in pentacene thin films deposited on Au(111).28,29 Therefore, we can deduce that in molecules containing strongly electronegative atoms the influence on the charge delocalization of the molecular backbone (pentacene versus perylene) and the different molecular arrangement (recumbent versus flat lying) may cause the difference observed when comparing energy dependent XPS spectra of H2Pc, PTCDA, and MOP.
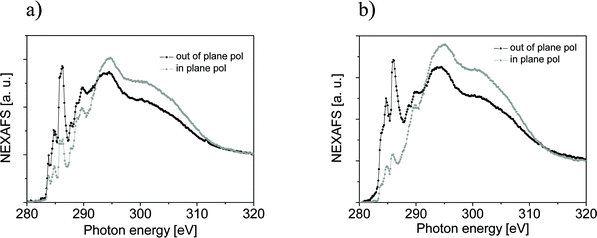 | ||
| Fig. 2 C1s NEXAFS spectra obtained from (a) a 106 Å MOP assembly and (b) a 76 Å MOPF assembly. The spectra were taken in grazing incidence for out-of-plane (black curve) and in-plane (grey curve) polarization. | ||
The comparison of the MOPF XPS spectra obtained at 330 and 1000 eV photon energies show a stronger difference in shape, indicating that the fluorine atoms play an even more pronounced role than the electronegativity in oxygen-containing molecules. This allows a deepening of our analysis including a quantitative point of view. The major difference in the two experiments is due to different surface (bulk) sensitivities: higher (lower) in the case of 330 eV (we can estimate a change in the inelastic mean free path from 4 to 15 Å, in the two cases25,26). Varying the surface sensitivity in a XPS experiment is, in fact, a way to investigate the surface core level shifts (SCLS), i.e., the photoemission binding energy difference of a core level of a surface with respect to a bulk atom/molecule.
SCLS have been found in various materials and are related to different origins in metals and semiconductors.30–34 In previous work25,26 we have analyzed the SCLS in PTCDA, coronene, and metal free phthalocyanines and no shift was observed, within an uncertainty of 0.1 eV.25,26,35 To explore the possibility of a SCLS, we have enhanced our fitting procedure by introducing two components, a bulk and a surface one for each contribution of the previous fit related to oxygen and fluorine atoms. By fitting the curves at 330 and 1000 eV, and keeping all the constraints we have previously used, we obtain a change in the surface/bulk components only for the C–F contribution (Fig. 1d, at 1000 eV the surface component decreases to 40% of its intensity at 330 eV, the bulk component shows the opposite behaviour). There is a small change in the C–O component similar to that observed in MOP, but no clear indication of a SCLS. Therefore, our fit would support the occurrence of a 0.25 eV SCLS, due to the presence of fluorine atoms: the difference observable in the energy dependent MOPF spectra can be explained, as in classical semiconductors, due to a redistribution of the charge on the surface atoms/molecules in the ground state because of the presence of the F atoms. In fact, this model has been successfully used, for example, for SCLS in III–V semiconductors, where the levels of the group III atoms at the surface show larger binding energies while those of the group V atoms are shifted to smaller binding energies because of a deviation of the charge distribution at the surface in comparison with the bulk.31,33
It is also important to mention that we performed the experiments on purpose far from the interface. The sampling depth ranges between 12 and 45 Å (corresponding to the estimated change in the inelastic mean free path from 4 to 15 Å). It means that the XPS spectra do not contain contributions from the interface. Furthermore, considering the nanorod morphology of the assemblies (see AFM images) we are undoubtedly far from the interface and we can exclude screening effects due to the substrate.
Fig. 1 shows also the O1s core level spectra of MOP and MOPF, and the MOPF F1s core level spectra. XPS O1s spectra present, as expected, only a main line: peaking at 533.14 eV in the MOP curves and at 532.89 eV in MOPF, and a small satellite feature at 1.25 and 0.95 eV (higher binding energy), for MOP and MOPF, respectively. F1s core level spectra show a main peak at 687.14 eV. The first HOMO–LUMO shake up satellite has a binding energy of 688.29 eV which is 1.15 eV away from the main line: the energy positions of the satellites in O1s and F1s spectra are in good agreement with the HOMO–LUMO satellite of the C1s spectra. The shoulder visible at around 531 eV is caused by a small fraction of impurities due to slight oxidation during handling and storage.
Our interpretation of the presented XPS data is based on a solid fit procedure cross-checked and supported by the available knowledge on core-level photoemission. However, in this framework, a detailed ab initio theoretical model of the photoemission events in organic thin films including intermolecular interactions, solid state effects, correlating structure with molecular packing and morphology, change in the Madelung constant, and charge delocalization would be very useful to further understand our experimental findings.
The XPS substrate signal attenuation during deposition offers the possibility to monitor the growth mode, as shown in Fig. 3. The obtained attenuation gives a clear indication for island formation. In this specific case, for both molecules, it is very difficult to estimate whether at least a complete first layer of molecules is deposited on the substrate. The very slow decay of the substrate intensity and its persistence also at high nominal thickness would point to the island (Volmer–Weber) growth mode, i.e., to a system formed only by nanorods.
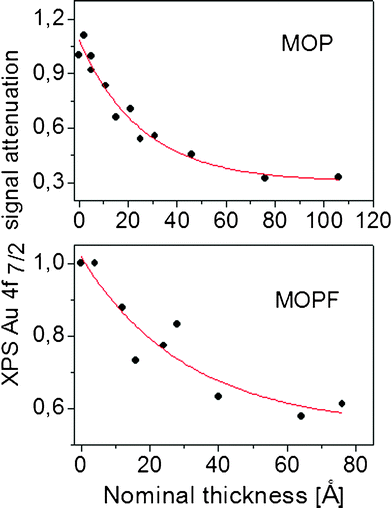 | ||
| Fig. 3 Relative Au4f XPS intensity as a function of assembly thickness. The solid line is a fit of the experimental data by using a first order exponential decay. | ||
To gain a deeper understanding, we have investigated the morphology by using AFM. Fig. 4 (upper panel) shows typical AFM pictures of samples after MOP or MOPF deposition, which exhibit images of nanorods of both molecules, confirming the island formation obtained by the XPS monitoring.
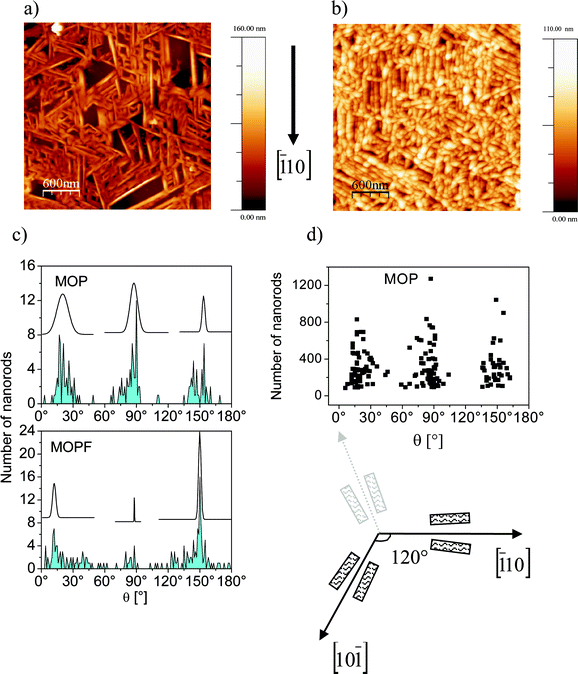 | ||
| Fig. 4 AFM images. Upper panel: 3 μm × 3 μm AFM images of (a) a 106 Å MOP assembly. (b) A 76 Å MOPF assembly. Lower panel: (c) angular distribution of the nanorods in MOP and MOPF together with the relative Gaussian curves as a visual guide. (d) Angular distribution of the nanorod length in MOP. A sketch of a possible nanorod growth scenario along the in-plane directions on a fcc(111) surface is also shown. The nanorods are sketched as rectangles. | ||
At a glance the rods seem to grow following specific directions. We have analyzed the images statistically and the results, obtained on more than 3500 nanorods, are shown in Fig. 4c and 4d. According to this analysis, MOP nanorods grow mainly along three directions forming an angle of 60° with each other. There is no preferential diffusion along one of these particular directions (we also verified the same behaviour for width and height of the nanorods of both molecules) as it may be deduced by the fact that the length of the rods is equally distributed along the three directions (Fig. 4d, Θ = 0° is an arbitrary direction).
A preferential growth direction of the rods is a phenomenon that is related to the threefold geometry of the Au(111) substrate. We have previously observed analogous morphology dependence on the substrate surface lattice in diindenoperylene thin films where the shape of the islands is directly related to the two-/three-fold geometry of the substrate.21,22,36 This effect is very well known for homoepitaxial and heteroepitaxial metal on metal growth.37
Very recently, analogous effects have been explored for needles of para-hexaphenyl and sexithiophene grown on muscovite and phlogopite mica.38 In particular, the higher symmetry of phlogopite mica leads to a triangular structure in the arrangement of the needles, as seen here for MOP on Au(111), while on muscovite the needles grow parallel.38,39 As discussed by Simbrunner et al.38 and also by Clancy from a more general perspective,40 this behaviour is based on the fact that a molecule prefers to adsorb with its long/short molecular axis with a certain azimuthal angle on the substrate. This angle depends on the surface–molecule combination.38–40
A similar analysis of the nanorods angular dependence in MOPF reveals a change: the symmetry is still related to the substrate, although with a different angular distribution (Fig. 4c, lower panel). Preliminary thickness dependent ultraviolet photoelectron spectroscopy investigations (not shown here) performed on the present nanorods show a change in the work function upon MOP and MOPF deposition on Au(111). The change is smaller in MOPF, pointing towards a behaviour similar to perfluoropentacene where the bonding distance of the molecules on Au(111)41 and Cu(111)42 surfaces is larger than for pentacene because of the push back effect due to the fluorine atoms.41,42 Thus, the MOPF bond to the substrate is weaker than for MOP; consequently the molecule–molecule interaction may be stronger for MOPF.
We also note that MOPF has a dipole moment, contrary to MOP. The presence of the dipole may further enhance the strength of the intermolecular interaction in comparison with MOP. According to the simulation proposed by Simbrunner et al.,38 the molecule–molecule interaction is responsible for a readjustment in the long molecular axis with respect to the substrate during nanorod growth. All these factors may cause MOPF molecules to preferentially adsorb with a different azimuthal angle with respect to the surface compared to MOP.
In particular, we note a preference for a parallel alignment of the nanorods, or for a 120° angle difference, as seen in large areas of the AFM images (Fig. 4b) as well as in the statistical angular distribution (Fig. 4c, lower panel). If we assume that the azimuthal angle of 120° describes the orientation of MOPF molecules on Au(111)(by a fortunate coincidence), we would expect nanorods only along directions with a difference of 120° between them due to the threefold geometry of the gold substrate (see sketch in Fig. 4). All directions with a smaller angle would be less favourable, as seen in the experiment (see Fig. 4).
Simbrunner et al.38 discuss the nanorod growth on mica in purely geometric terms, excluding that electric fields may be the driving force for needle growth. This model seems to have a general relevance, and it may be also useful on metal substrates, as our work shows. However, the comparison between the MOP and MOPF angular distribution of the nanorods also points to the necessity to refer to the polar/non polar nature of the molecules.
Conclusions
In conclusion, the richness of phenomena we have found in the electronic structure of the investigated newly synthesized substituted pentacenes underlines that their electronic structure is strongly affected, on one side by the chosen substituents, on the other, by the interaction of the molecules with the surrounding environment.The template effect of the substrate geometry is a key that can be used in device engineering: by decreasing the symmetry of the substrate lattice we expect to obtain aligned nanorods that can be used in interdigitated device configurations.
We believe that our experimental work is a step forward in the comprehension of this class of material and may stimulate the necessary ab initio calculations to shed light on the complex mechanisms competing in the screening of the core–hole in photoemission. We also underline that the presence of fluorine atoms not only strongly influences the electronic structure of the molecules as found in previous work,41,42 but also the assembly morphology and the distribution on the substrate. This is a relevant result for applications in electronics, given the importance of fluorination in the optimization of molecules for use in devices like junctions or photovoltaic cells.
Acknowledgements
The authors thank the Helmholtz-Zentrum Berlin (HZB) electron storage ring BESSY II, for providing beamtime, HZB resident staff, in particular Dr S. Krause for beamtime support; and W. Neu, University of Tübingen, for technical support. Financial support from the Helmholtz-Zentrum Berlin is gratefully acknowledged.References
- D. Braga and G. Horowitz, Adv. Mater., 2009, 21, 1473 CrossRef CAS.
- C. W. Tang and S. A. Van Slyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS.
- S. R. Forrest, Org. Electron., 2003, 4, 45 CrossRef.
- F. Garnier, A. Yassar, R. Hajlaoui, G. Horowitz, F. Dloffre, B. Servet, S. Ries and P. Alnot, J. Am. Chem. Soc., 1993, 115, 8716 CrossRef CAS.
- C. D. Dimitrakopoulos and P. R. L. Malefant, Adv. Mater., 2002, 14, 99 CrossRef CAS.
- J. E. Anthony, Chem. Rev., 2006, 106, 5028 CrossRef CAS.
- C. Tönshoff and H. F. Bettinger, Chem.–Eur. J., 2012, 18, 1789 CrossRef.
- Y. Sakamoto, T. Suzuki, M. Kobayashi, Y. Gao, Y. Fukai, Y. Inoue, F. Sato and S. Tokito, J. Am. Chem. Soc., 2004, 126, 8138 CrossRef CAS.
- Z. Cheng, E. S. Chu, D. Sun, D. Kim, Y. Zhu, M. Luo, G. Pawin, K. L. Wong, K.-Y. Kwon, R. Carp, M. Marsella and L. Bartels, J. Am. Chem. Soc., 2010, 132, 13578 CrossRef CAS.
- M. Schiek, F. Balzer, K. Al-Shamery, A. Lützen and H.-G. Rubahn, J. Phys. Chem., 2009, 113, 9601 CAS.
- R. Hesse, T. Chassé, P. Streubel and R. Szargan, Surf. Interface Anal., 2004, 36, 1373 CrossRef CAS.
- A. Schöll, Y. Zou, D. Huebner, S. G. Urquhart, Th. Schmidt, R. Fink and E. Umbach, J. Chem. Phys., 2005, 123, 044509 CrossRef.
- A. Schöll, Y. Zou, M. Jung, Th. Schmidt, R. Fink and E. Umbach, J. Chem. Phys., 2004, 121, 10260 CrossRef.
- C. Baldacchini, F. Allegretti, R. Gunnella and M. G. Betti, Surf. Sci., 2007, 601, 2603 CrossRef CAS.
- M. Alagia, C. Baldacchini, M. G. Betti, F. Bussolotti, V. Carravetta, U. Ekström, C. Mariani and S. Stranges, J. Chem. Phys., 2005, 122, 124305 CrossRef.
- J. B. Gustafsson, H. M. Zhang, E. Moons and L. S. O. Johansson, Phys. Rev. B, 2007, 75, 155413 CrossRef.
- G. Gravila, D. R. T. Zahn and W. Braun, Appl. Phys. Lett., 2006, 89, 162102 CrossRef.
- A. Nikitin, H. Ogasawara, D. Mann, R. Denecke, Z. Zhang, H. Dai, K. Cho and A. Nilsson, Phys. Rev. Lett., 2005, 95, 225507 CrossRef CAS.
- M. L. M. Rocco, M. Haeming, D. R. Batchelor, R. Fink, A. Schöll and E. Umbach, J. Chem. Phys., 2008, 129, 074702 CrossRef CAS.
- B.-E. Schuster, M. B. Casu, I. Biswas, A. Hinderhofer, A. Gerlach, F. Schreiber and T. Chassé, Phys. Chem. Chem. Phys., 2009, 11, 9000 RSC.
- M. B. Casu, S.-A. Savu, P. Hoffmann, B.-E. Schuster, O. Mentes, M. A. Niño, A. Locatelli and T. Chassé, CrystEngComm, 2011, 13, 4139 RSC.
- M. B. Casu, B.-E. Schuster, I. Biswas, C. Raisch, H. Marchetto, Th. Schmidt and T. Chassé, Adv. Mater., 2010, 22, 3740 CrossRef CAS.
- E. E. Rennie, B. Kempgens, H. M. Köppe, U. Hergenhahn, J. Feldhaus, B. S. Itchkawitz, A. L. D. Kilcoyne, A. Kivimäki, K. Maier, M. N. Piancastelli, M. Polcik, A. Rüdel and A. M. Bradshaw, J. Chem. Phys., 2000, 113, 7362 CrossRef CAS.
- K. Ueda, M. Hoshino, T. Tanaka, M. Kitajima, H. Tanaka, A. De Fanis, Y. Tamenori, M. Ehara, F. Oyagi, K. Kuramoto and H. Nakatsuji, Phys. Rev. Lett., 2005, 94, 243004 CrossRef.
- M. B. Casu, Y. Zou, S. Kera, D. Batchelor, Th. Schmidt and E. Umbach, Phys. Rev. B, 2007, 76, 193311 CrossRef.
- M. B. Casu, Phys. Status Solidi RRL, 2008, 2, 40 CrossRef CAS.
- J. Stöhr, NEXAFS Spectroscopy, Springer-Verlag, Berlin, Heidelberg, New York, 1998 Search PubMed.
- D. Käfer, L. Ruppel and G. Witte, Phys. Rev. B, 2007, 75, 085309 CrossRef.
- R. A. Cantrell, C. James and P. Clancy, Langmuir, 2011, 27, 9944 CrossRef CAS.
- S. F. Alvarado, M. Campagna and W. Gudat, J. Electron Spectrosc. Relat. Phenom., 1980, 18, 43 CrossRef CAS.
- D. E. Eastman, T.-C. Chiang, P. Heimann and F. J. Himpsel, Phys. Rev. Lett., 1980, 45, 656 CrossRef CAS.
- R. E. Watson, J. W. Davenport, M. L. Perlman and T. K. Sham, Phys. Rev. B, 1981, 24, 1791 CrossRef CAS.
- W. F. Egelhoff Jr., Surf. Sci. Rep., 1987, 6, 253 CrossRef.
- R. E. Watson and J. W. Davenport, Phys. Rev. B, 1983, 27, 6418 CrossRef CAS.
- D. Kolacyak, H. Peisert and T. Chassé, Appl. Phys. A, 2009, 95, 173 CrossRef CAS.
- M. B. Casu, Cryst. Growth Des., 2011, 11, 3629 CAS.
- H. Brune, Surf. Sci. Rep., 1998, 31, 121 CAS.
- C. Simbrunner, D. Nabok, G. Hernandez-Sosa, M. Oehzelt, T. Djuric, R. Resel, L. Romaner, P. Puschnig, C. Ambrosch-Draxl, I. Salzmann, G. Schwabegger, I. Watzinger and H. Sitter, J. Am. Chem. Soc., 2011, 133, 3056 CrossRef CAS.
- F. Balzer, M. Schiek, H.-G. Rubahn, K. Al-Shamery and A. Lutzen, J. Vac. Sci. Technol. B, 2008, 26, 1619 CAS.
- P. Clancy, Chem. Mater., 2011, 23, 522 CrossRef CAS.
- N. Koch, A. Vollmer, S. Duhm, Y. Sakamoto and T. Suzuki, Adv. Mater., 2007, 19, 112 CrossRef CAS.
- K. Toyoda, I. Hamada, S. Yanagisawa and Y. Morikawa, Org. Electron., 2011, 12, 295 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |