An organosilane route to mesoporous silica nanoparticles with tunable particle and pore sizes and their anticancer drug delivery behavior†
Wenru
Zhao
a,
Hongti
Zhang
a,
Shu
Chang
a,
Jinlou
Gu
a,
Yongsheng
Li
a,
Liang
Li
a and
Jianlin
Shi
*ab
aLow dimensional Materials Chemistry Laboratory, School of Materials Science and Engineering, East China University of Science and Technology, Shanghai 200237, China
bState Key Laboratory of High Performance Ceramics and Superfine Microstructures, Shanghai Institute of Ceramics, Chinese Academy of Sciences, 1295 Ding-xi Road, Shanghai 200050, China. E-mail: jlshi@sunm.shcnc.ac.cn
First published on 11th April 2012
Abstract
Uniform mesoporous silica nanospheres were synthesized using organosilane as a template. By adjusting the water/ethanol ratio and the amount of the silica precursors, the pore and particle sizes can be tuned in a certain range. The materials show a sustained and pH-sensitive drug release property and a comparable antitumor efficacy.
In recent years, mesoporous silica nanospheres (MSNs) have been well developed as effective drug storage vehicles in drug delivery systems1 owing to their large pore volume, high surface area,2 ease of functionalization,3 low toxicity and biodegradability.4 For their application in drug delivery, the morphology control of MSNs, especially their particle size, dispersivity and pore size are important issues because particles or aggregates with sizes above 300 nm may lead to thrombosis,5 and the pore diameter determines the dimensions of drug molecules which can be loaded in them.
Various approaches have been developed for the preparation of MSNs, including self-assembly,6 emulsions,7 and post-etching,8etc. Among them, synergetic self-assembly between a cationic surfactant, e.g. hexadecyltrimethyl ammonium bromide, and a silica precursor, e.g. tetraethyl orthosilicate (TEOS), has been proven to be an effective method to fabricate uniform spherical MCM-41 nanoparticles which are the typical MSNs for the exploration of MSNs as sustained/controlled drug delivery carriers.9 Although the particle diameter and pore size of such MCM-41-type MSNs can be tuned through changing some synthetic conditions,10 they are difficult to adjust independently. Furthermore, the morphology and structure of such MSNs are extremely sensitive to the synthetic conditions,11 such as concentration and temperature. Therefore, it is of great significance to develop new and effective methods to fabricate MSNs with tunable particle diameter and pore size.
In recent years, a novel type of submicrometer-sized mesoporous silica spheres with a core-shell structure employing long chain organosilane as a porogen instead of the traditional cationic surfactants has been reported by several groups.12 The organosilane and TEOS hydrolyze and subsequently coat together on the pre-prepared cores, forming an organic–inorganic hybrid shell. One advantage of this method is that the multi-silica precursors can co-condense simultaneously, which leads to the easy control of the final core-shell structure and dispersivity. However, the study of the microstructure synthesized by this method, especially on the pore diameter and particle size, is still limited. Herein, we successfully synthesized a kind of uniform MSN through the co-condensation of a long chain organosilane, n-octadecyltrimethoxysilane (C18TMS), and TEOS. The particle size can be tuned in the range of 80–300 nm by changing the amount of multi-silica precursors. The pore diameter of such MSNs can be regulated from 2.0 to 4.6 nm by adjusting the molar ratio of water to ethanol. Unlike MCM-41-type MSNs, the particle and pore size in this case can be tuned independently. In the present work, doxorubicin (DOX), a typical antitumor drug, was chosen to be stored in the channels of the MSNs. The in vitro experimental results show drug storage and release, biocompatibility and antitumor efficacy.
The preparation of size tunable MSNs is based on the traditional silicate sol–gel approach. The silica precursors, C18TMS and TEOS, were firstly mixed at the molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.7 and dropped slowly into the starting reaction solution containing H2O, ethanol, and ammonia under vigorous stirring, followed by reaction for 4 h at ambient temperature. The resultant C18TMS-incorporated particles were centrifuged, washed by ethanol, dried, and calcined to remove organic groups at 550 °C for 6 h. The experimental details are listed in Table S1, see ESI.†
:4.7 and dropped slowly into the starting reaction solution containing H2O, ethanol, and ammonia under vigorous stirring, followed by reaction for 4 h at ambient temperature. The resultant C18TMS-incorporated particles were centrifuged, washed by ethanol, dried, and calcined to remove organic groups at 550 °C for 6 h. The experimental details are listed in Table S1, see ESI.†
The typical TEM and SEM images of the MSNs (sample B2 in Table S1†) are presented in Fig. 1 (a, b, c). It can be seen that all the particles are spherical, well dispersed and uniform in particle size of around 150 nm. The worm-like mesopores arrange randomly. No obvious diffraction peak appears in the low angle XRD pattern (Fig. S1†) indicating a poorly aligned mesoporous structure. The mesoporous structure is further confirmed by the N2 adsorption–desorption measurements, which exhibit type IV isotherms with a leap at relative pressure of 0.4 and an H3 hysteresis loop (Fig. 1d). The corresponding pore diameter distribution is narrow and centered at around 4.4 nm. The obtained MSNs have a specific surface area of 552 m2g−1 and a pore volume of 0.54 cm3g−1.
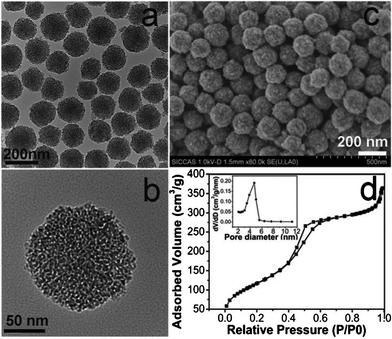 | ||
| Fig. 1 TEM (a, b), SEM (c) images and N2 adsorption-desorption isotherms (d) of the typical MSNs (sample B2). Inset in (d) is the corresponding pore size distribution. | ||
The pore size control in this work was achieved just by adjusting the H2O/EtOH ratios in the starting suspensions. The N2 adsorption–desorption isotherms (Fig. 2A) of all the MSNs exhibit type IV features demonstrating their mesoporous structures. when the ratio of H2O/EtOH increases from 1:3.86 to 1:1.24, the average pore diameters can be enlarged from 2.3 to 4.6 nm (Fig. 2B and Table S1, see ESI†), which may be attributed to the C18TMS micellar size. During the reaction, there is a competition between H2O and ethanol in the starting solutions. The existence of H2O enlarges the hydrophobic C18TMS micelles. Meanwhile, ethanol weakens the aggregation of C18TMS and decreases the size of micelles. Therefore, the size of C18TMS micelles, which determines the pore diameter, is very sensitive to the molar ratio of H2O/EtOH. However, from the TEM images (Fig. S2†), it can be seen that the optimal ratios of H2O/EtOH are from 1:2.29 to 1:1.24 to keep the high dispersivity of the MSNs.
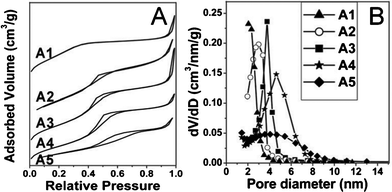 | ||
| Fig. 2 N2 adsorption-desorption isotherms (A) and the corresponding pore diameter distributions (B) of MSNs synthesized at H2O:EtOH molar ratios of 1:3.86 (A1), 1:2.29 (A2), 1:1.78 (A3), 1:1.24 (A4) and 1:0.77 (A5), respectively. | ||
For the synthesis of uniform MSNs with controlled particle sizes, we changed the amount of the TEOS and C18TMS mixture while fixing other synthetic conditions. TEM images (Fig. 3 B1–B4) shows that, with the increasing amount of TEOS and C18TMS mixture, the mean diameters of MSNs became larger gradually while keeping the good dispersivity of the particles. Fig. 3B gives the particle size distributions which were calculated from the corresponding TEM image by counting more than 100 particles. The particle size distributions are sharp and the mean sizes of MSNs can be tuned from 80 nm to 330 nm in Fig. 3B, which is consistent with the corresponding dynamic light scattering result (Fig. S3, ESI†). This can be mainly due to the double roles of C18TMS, which is not only a porogen, but also a silicate source. During the formation of MSNs, C18TMS is involved in the hydrolysis and subsequent co-condensation with TEOS. The growth of the mesoporous spheres follows a similar process to that of silica spheres synthesized by the Stöber method,13 in which the silicate source hydrolyzes and condenses, forming monodispersed silica spheres. The dynamic light scattering result (Fig. S3†) shows that the particle size distributions are wider than the TEM result, indicating that some aggregation occurred. Additionally, it is worth mentioning that the pore size, surface area and pore volume of the MSNs are not influenced significantly by the growth process of the MSNs (Table S1†), indicating that the mesoporous structure of such MSNs is independent of the amount of multi-silica precursors.
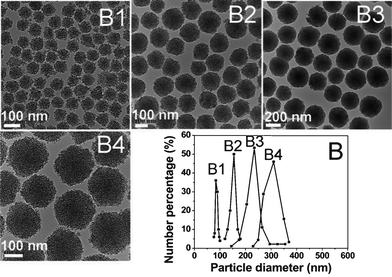 | ||
| Fig. 3 TEM images (B1, B2, B3, B4) and corresponding particle size distributions (B) of MSNs fabricated by adding 200 (B1), 400 (B2), 650 (B3), 900 (B4) μl of the TEOS/C18TMS mixture to 48.2 ml of the starting solution, respectively. | ||
Considering the smallest particle size and the good mesoporous properties, we chose sample B1 to investigate the feasibility of the as-synthesized MSNs acting as drug loading and releasing vehicles.
The storage of DOX in mesoporous channels can be assessed by N2 adsorption–desorption isotherms and FTIR spectra. After the storage of DOX, the N2 adsorbed volumes obviously decrease while the isotherms remain as Langmuir type IV, implying that the mesopores have been partly occupied by DOX molecules (Fig. S4, ESI†). The same result can also be concluded from the corresponding mesoporous structural parameters, such as pore volume (0.32 cm3 g−1) and surface area (216.9 m2 g−1) of DOX-loaded MSNs, which are almost half of the original data of sample B1. In the FTIR spectra (Fig. S5, ESI†), after DOX storage, an aromatic –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– stretching vibration band at the wave number around 1500 cm−1 is present and the Si–OH band at 960 cm−1 still remains, demonstrating that DOX molecules have been stored in MSNs physically without interacting with the Si–OH groups on the pore surface. The DOX loading capacity of MSNs was determined to be 162 mg g−1 by UV absorbance spectroscopy at 490 nm, the characteristic wavelength of DOX.
C– stretching vibration band at the wave number around 1500 cm−1 is present and the Si–OH band at 960 cm−1 still remains, demonstrating that DOX molecules have been stored in MSNs physically without interacting with the Si–OH groups on the pore surface. The DOX loading capacity of MSNs was determined to be 162 mg g−1 by UV absorbance spectroscopy at 490 nm, the characteristic wavelength of DOX.
Fig. S6† shows the DOX release behaviors in vitro from the DOX-loaded MSN system over a 160 h period in PBS solutions at pH 4.4, 5.4, and 6.4. Apparently, the DOX release rate is pH-dependent. It can be seen that a burst release occurred within the first 20 h from the DOX-loaded MSNs systems at different pH values. In a pH 6.4 solution, the release reached its maximum (25%) in 30 h. In a more acidic solution of pH = 5.4, the release amount reached 50% in 60 h. Under strong acidic conditions, pH = 4.4, after a fast release within the first 20 h, a sustained release followed and 70% of DOX released in 100 h. This pH-dependent release character is very important and useful because the pH value can be significantly lowered in tumor cells by/during the particle endocytosis compared to the near-neutral conditions outside of cells.
To reveal the uptake of the nanoparticles by human breast cancer MCF-7 cells and the intracellular location, bio-TEM analysis was carried out on the ultrathin section of MCF-7 cells after the incubation with the nanoparticles. The distribution of MSNs can be clearly observed in bio-TEM images (Fig. 4a), and a large number of them are found in the cytoplasm as marked by red circle (Fig. 4b) while no nanocarriers are found in the nucleus. In addition, some of the nanoparticles can be found near the cell membrane, forming vesicles (as marked by black circle, Fig. 4c). After uptake by cancer cells via endocytosis, the nanoparticles will be processed in endosomes and lysosomes, and the DOX molecules will eventually release into the cytoplasm14 of a relatively acidic environment. To verify whether the released DOX from MSNs is pharmacologically active, the cytotoxic effect of the DOX-loaded MSNs on MCF-7 cells was tested. As shown in Fig. S7, see ESI,† DOX-loaded MSNs caused high amounts of MCF-7 cell death similar to free DOX, indicating that the DOX delivered by MSNs retained its pharmaceutical activity. The cytotoxicity against MCF-7 cells demonstrates an incubating time and concentration dependency. Meanwhile, the MSN cargo itself shows no visible cytotoxicity against MCF-7 cells (Fig. S8A†) as well as representative normal cells, fibroblast L929 cells (Fig. S8B†).
 | ||
| Fig. 4 The Bio-TEM images of MCF-7 cells after incubation with MSNs for 24 h (a). (b) and (c) are the magnifications of the red (b) and black (c) circled sections in (a), respectively. | ||
In summary, we have successfully synthesized uniform MSN using a long chain organosilane, C18TMS, as both pore template and silica source co-condensing with TEOS. The particle size can be tuned in the range of 80–300 nm by changing the amount of multi-silica precursors, while the pore diameter can be regulated from 2.0 to 4.6 nm by adjusting the molar ratio of water to ethanol. The synthesized MSNs showed very low cytotoxicity towards human breast cancer MCF-7 cells and normal fibroblast L929 cells. An anticancer drug, DOX, can be effectively loaded into the pore channels of MSNs, which exhibits a pH-dependent release property in vitro and high cytotoxicity comparable to free DOX. All these positive results make MSNs a promising candidate for drug delivery applications. The MSNs may be further conjugated with some functional materials (e.g. magnetic or fluorescent nanoparticles) to construct multifunctional systems for the diagnosis and targeted therapy of cancers.
This work was supported by the National Basic Research Program of China (973 Program, 2012CB933602), the National Natural Science Foundation of China (Grant No. 21001043), the Natural Science Foundation of Shanghai (Grant No. 10ZR1407500), the Fundamental Research Funds for the Central Universities (Grant No. WD0911012 and WD1114004), and the Program for Changjiang Scholars and Innovative Research Team in University (Grant No. IRT0825).
References
- (a) S. Angelos, M. Liong, E. Choi and J. I. Zink, Chem. Eng. J., 2008, 137, 4 CrossRef CAS; (b) J. L. Vivero-Escoto, I. I. Slowing, B. G. Trewyn and V. S. Y. Lin, Small, 2010, 6, 1952 CrossRef CAS.
- M. Vallet-Regi, A. Ramila, R. P. del Real and J. Perez-Pariente, Chem. Mater., 2001, 13, 308 CrossRef CAS.
- C. H. Lei, P. Liu, B. W. Chen, Y. M. Mao, H. Engelmann, Y. Shin, J. Jaffar, I. Hellstrom, J. Liu and K. E. Hellstrom, J. Am. Chem. Soc., 2010, 132, 6906 CrossRef CAS.
- Q. J. He, Z. W. Zhang, Y. Gao, J. L. Shi and Y. P. Li, Small, 2009, 5, 2722 CrossRef CAS.
- C. B. Barbé, J. Bartlett, L. Kong, K. Finnie, H. Q. Lin, M. Larkin, S. Calleja, A. Bush and G. Calleja, Adv. Mater., 2004, 16, 1959 CrossRef.
- (a) S. Gai, P. Yang, P. Ma, D. Wang, C. Li, X. Li, N. Niu and J. Lin, J. Mater. Chem., 2011, 21, 16420 RSC; (b) T. Yokoi, T. Karouji, S. Ohta, J. N. Kondo and T. Tastsumi, Chem. Mater., 2010, 22, 3900 CrossRef CAS; (c) Q. He, J. Shi, X. Cui, C. Wei, L. Zhang, W. Wu, W. Bu, H. Chen and H. Xu, Chem. Commun., 2011, 47, 7947 RSC.
- (a) H. Zhang, Z. Li, P. Xu, R. Wu and Z. Jiao, Chem. Commun., 2010, 46, 6783–6785 RSC; (b) C. I. Zoldes and A. Imhof, Adv. Mater., 2005, 17, 924 CrossRef.
- Q. Zhang, T. Zhang, J. Ge and Y. Yin, Nano Lett., 2008, 8, 2867 CrossRef CAS.
- (a) J. E. Lee, N. Lee, H. Kim, J. Kim, S. H. Choi, J. H. Kim, T. Kim, I. C. Song, S. P. Park, W. K. Moon and T. Hyeon, J. Am. Chem. Soc., 2010, 132, 552 CrossRef CAS; (b) L-S. Wang, L-C. Wu, S-Y. Lu, L-L. Chang, I-T. Teng, C-M. Yang and J. A. Ho, ACS Nano, 2010, 4, 4371 CrossRef CAS; (c) C-H. Lee, S-H. Cheng, I-P. Huang, J. S. Souris, C-S. Yang, C-Y. Mou and L-W. Lo, Angew. Chem., Int. Ed., 2010, 49, 8214 CrossRef CAS.
- I. I. Slowing, J. L. Vivero-Escoto, C. W. Wu and V. S. Y. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278 CrossRef CAS.
- (a) S. Huh, J. W. Wiench, J-C. Yoo, M. Pruski and V. S. Y. Lin, Chem. Mater., 2003, 15, 4247 CrossRef CAS; (b) Z. Qiao, L. Zhang, M. Guo, Y. Liu and Q. Huo, Chem. Mater., 2009, 21, 3823 CrossRef CAS; (c) I. I. Slowing, B. G. Trewyn and V. S. Y. Lin, J. Am. Chem. Soc., 2007, 129, 8845 CrossRef CAS; (d) T. Suteewong, H. Sai, R. Cohen, S. Wang, M. Bradbury, B. Baid, S. M. Gruner and U. Wiesner, J. Am. Chem. Soc., 2011, 133, 172 CrossRef CAS.
- (a) G. Büchel, K. K. Unger, A. Matsumoto and K. Tsutsumi, Adv. Mater., 1998, 10, 1036 CrossRef; (b) W. Zhao, J. Gu, L. Zhang, H. Chen and J. Shi, J. Am. Chem. Soc., 2005, 127, 8916 CrossRef CAS; (c) W. Zhao, H. Chen, Y. Li, L. Li, M. Lang and J. Shi, Adv. Funct. Mater., 2008, 18, 2780 CrossRef CAS; (d) Y. Chen, H. R. Chen, L. M. Guo, Q. J. He, F. Chen, J. Zhou, J. W. Feng and J. L. Shi, ACS Nano, 2010, 4, 529 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62 CrossRef.
- (a) Y. Chen, H. Chen, D. Zeng, Y. Tian, F. Chen, J. Feng and J. Shi, ACS Nano, 2010, 4, 6001 CrossRef CAS; (b) J. Gu, S. Su, Y. Li, Q. He, J. Zhong and J. Shi, J. Phys. Chem. Lett., 2010, 1, 3446 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra20166f/ |
| This journal is © The Royal Society of Chemistry 2012 |