Monodisperse bifunctional Fe3O4@NaGdF4:Yb/Er@NaGdF4:Yb/Er core–shell nanoparticles†
Chongna
Zhong
a,
Piaoping
Yang
*a,
Xingbo
Li
a,
Chunxia
Li
b,
Dong
Wang
a,
Shili
Gai
a and
Jun
Lin
*b
aKey Laboratory of Superlight Materials and Surface Technology, Ministry of Education, Harbin Engineering University, Harbin, China. E-mail: yangpiaoping@hrbeu.edu.cn
bState Key laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, China. E-mail: jlin@ciac.jl.cn
First published on 2nd February 2012
Abstract
We report for the first time the synthesis of monodisperse, magnetic and up-conversion luminescent Fe3O4@NaGdF4:Yb/Er@NaGdF4:Yb/Er core–shell structured nanoparticles (NPs) by a seed-growth process. The growth of a NaGdF4:Yb/Er shell on Fe3O4@NaGdF4:Yb/Er NPs can significantly enhance the up-conversion emission intensity.
In recent years, core–shell structured NPs have attracted extensive interest,1 because the combined functionalities of cores and shells endow them with great potential application in various fields.2 In particular, core–shell structured NPs with magnetism and fluorescence have been intensively pursued for their biomedical applications, such as contrast enhancement agents in magnetic resonance imaging (MRI), magnetic carriers for drug delivery,3 biological labeling,4 biological luminescent probes.5 However, conventional fluorescent organic dyes used in these core–shell composites are not suitable for sensitive detection and real-time monitoring because of the serious photobleaching,6 and quantum dots (QDs) generally suffer from the inherent toxicity.7 These drawbacks dramatically limit their biological applications. Rare earth based luminescent NPs show superior luminescent and chemical properties, such as high fluorescent efficiency,8 multicoloured emissions,9 high resistance to photobleaching, and especially low toxicity,10 making them highly suitable as alternatives to organic dyes and QDs in biological applications.11
Despite the considerable potential of nanomaterials for applications to bioscience and other fields,12 improvements are still needed to optimize the properties for potential commercial applications.13 One strategy to improve the luminescence efficiency of NPs is the use of core–shell architectures where a shell of material with similar lattice constants is grown around the luminescent nanoparticles to avoid the formation of defects at the core–shell interface.14 In this structure, the increase of the distance between the luminescent rare earth ions and the surface results in reducing the non-radiative pathway, and the quantum yield of the nanomaterials is consequently enhanced. Nowadays, many research teams are devoted to synthesizing this kind of nanocomposite.15 However, to the best of our knowledge, monodisperse core–shell NPs with magnetic and up-conversion luminescence have never been reported so far. Thus, designing an effective strategy to synthesize high-quality core–shell structured colloidal NPs is of significant value.
Herein, we first report a seed-growth procedure to synthesize a new type of core–shell structured NPs composed of magnetite core and up-conversion luminescent shell. Each of the components in the nanocomposites displays a unique function. Fe3O4 as the core endows nanocrystals with magnetic property, and the outer NaGdF4:Yb/Er shell is to improve the luminescent efficiency. This straightforward route may be applicable to the preparation of other bifunctional core–shell NPs.
The Fe3O4@NaGdF4:Yb/Er and Fe3O4@NaGdF4:Yb/Er@NaGdF4:Yb/Er core–shell NPs were synthesized by a modified high-temperature thermolysis process.16 The synthesis process is presented in Scheme 1. Oleic acid (OA) capped Fe3O4 core NPs were obtained from the thermal decomposition of the iron-oleate precursors. The as-prepared Fe3O4 NPs then served as the seeds for the shell growth in the presence of oleic acid and 1-octadecene, resulting in the formation of the core–shell NPs. A luminescence shell was further grown using the as-prepared Fe3O4@NaGdF4:Yb/Er as seeds. Oleic acid was selected as the surfactant to control the particle growth and to prevent the NPs from aggregation. The detailed experimental procedures are given in the experimental section.†
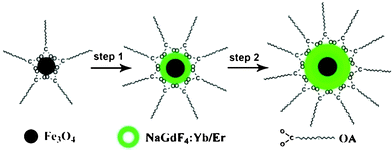 | ||
| Scheme 1 Schematic formation processes of Fe3O4@NaGdF4:Yb/Er@NaGdF4:Yb/Er NPs. | ||
Fig. 1 shows the transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images of as-prepared pure Fe3O4, Fe3O4@NaGdF4:Yb/Er, and Fe3O4@NaGdF4:Yb/Er@NaGdF4:Yb/Er, respectively. In Fig. 1A, Fe3O4 seeds consist of monodisperse NPs with narrow size distribution (inset in Fig. 1A). Fig. 1B clearly reveals a highly ordered nanoarray made up of circular NPs with an average diameter of 9 nm. In Fig. 1C, monodisperse Fe3O4@NaGdF4:Yb/Er core–shell structured NPs with a nearly spherical morphology are obtained and the average particle size is about 22 nm, which is much bigger than that of Fe3O4 NPs, suggesting that a NaGdF4:Yb/Er shell is successfully grown on the surface of Fe3O4 nanospheres. Moreover, there is no obvious boundary between the shell and core, which may be due to the ultra-small sizes of the Fe3O4 core NPs. The obvious lattice fringes in the HRTEM image (Fig. 2D) indicate the crystalline nature of Fe3O4@NaGdF4:Yb/Er NPs. The distances of 0.30 nm between the adjacent lattices agree well with the d110 spacing (0.301 nm) of hexagonal-phased NaGdF4 (JCPDS no. 27-0699). In Fig. 1E, it is found that the growth of further shell results in an obvious increase of the NPs size. The average particle diameter increases from 22 nm for Fe3O4@NaGdF4:Yb/Er NPs to 32 nm for Fe3O4@NaGdF4:Yb/Er@NaGdF4:Yb/Er core–shell NPs. Similarly, the particle sizes are uniform with good monodispersity and narrow size distribution (inset in Fig. 1E). The HRTEM image (Fig. 1F) clearly reveals the well-resolved lattices fringes, indicating the high crystallinity of the as-prepared sample, which are well consistent with the XRD results (Fig. S1 in the ESI†). The distance of the adjacent lattice fringes is determined to be about 0.30 nm, which corresponds well to the d110 spacing of hexagonal phase NaGdF4.
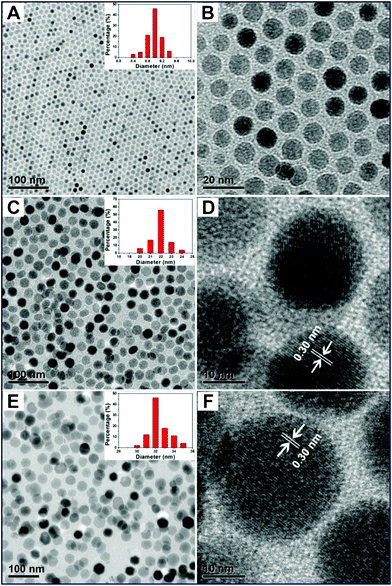 | ||
| Fig. 1 (A) Low-resolution, (B) high-resolution TEM image of Fe3O4 NPs; (C) low-resolution, (D) high-resolution TEM images of Fe3O4@NaGdF4:Yb/Er NPs; (E) low-resolution, (F) high-resolution TEM images of Fe3O4@NaGdF4:Yb/Er@NaGdF4:Yb/Er core–shell NPs. Insets are their corresponding particle size distribution histograms. | ||
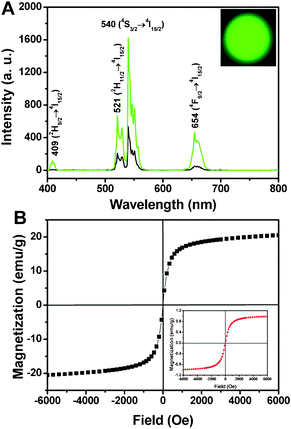 | ||
| Fig. 2 Up-conversion emission spectra of (A) Fe3O4@NaGdF4:Yb/Er (black line), Fe3O4@NaGdF4:Yb/Er@NaGdF4:Yb/Er (green line) and the corresponding photograph under 980 nm irradiation (inset); (B) magnetizations of pure Fe3O4 and Fe3O4@NaGdF4:Yb/Er@NaGdF4:Yb/Er NPs (inset) as a function of applied magnetic field measured at room temperature. | ||
The oleic acid (OA) plays an important role in controlling the particle growth, which is confirmed by the FT-IR spectrum, as shown in Fig. S2.† The absorption peaks at 2924 cm−1 and 2852 cm−1 can be ascribed to the asymmetric (νas) and symmetric (νs) stretching vibration of methylene (–CH2) group. The peak at 1698 cm−1, 1567 cm−1 and 1467 cm−1 can be assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration, asymmetric (νas) and symmetric (νs) stretching vibrations of –COOH,17 respectively. The peak at 1112 cm−1 can be due to the C–F stretching vibration. The peak at 721 cm−1 is assigned to the in-planar swing of (CH2) n (n > 4).18 The EDS spectrum of Fe3O4@NaGdF4:Yb/Er@NaGdF4:Yb/Er core–shell NPs (Fig. S3 in the ESI†) confirms the presence of element F, O, Na, Gd, and Yb in the product. While the absence of element Fe may be due to the coated layer on the surface of Fe3O4 core.
O stretching vibration, asymmetric (νas) and symmetric (νs) stretching vibrations of –COOH,17 respectively. The peak at 1112 cm−1 can be due to the C–F stretching vibration. The peak at 721 cm−1 is assigned to the in-planar swing of (CH2) n (n > 4).18 The EDS spectrum of Fe3O4@NaGdF4:Yb/Er@NaGdF4:Yb/Er core–shell NPs (Fig. S3 in the ESI†) confirms the presence of element F, O, Na, Gd, and Yb in the product. While the absence of element Fe may be due to the coated layer on the surface of Fe3O4 core.
Fig. 2 shows the up-conversion emission spectra of the Fe3O4@NaGdF4:Yb/Er, Fe3O4@NaGdF4:Yb/Er@NaGdF4:Yb/Er and its corresponding photograph (inset) under 980 laser diode excitation, respectively. In both spectra, the typical emissions at 409, 521, 540, and 654 nm can be ascribed to the respective 2H9/2 → 4I15/2, 2H11/2 → 4I15/2, 4S3/2 → 4I15/2 and 4F9/2 → 4I15/2 transitions of Er3+ ions.19 The green emission at 540 nm is much stronger than that of the red one (654 nm), resulting in the bright green emission (inset in Fig. 2A). Moreover, it should be noted that the emission intensity of the Fe3O4@NaGdF4:Yb/Er@NaGdF4:Yb/Er NPs (green line in Fig. 2A) is almost four times to that of Fe3O4@NaGdF4:Yb/Er (black line), which may be caused by the significant elimination of non-radiative centers on the surface of Fe3O4@NaGdF4:Yb/Er NPs by the shielding effect of protective shell. In addition, the increased emission intensity may also derive from the suppressed quenching by the outer shell and the inherent emission of luminescent NaGdF4:Yb/Er shell. The magnetic hysteresis loops of pure Fe3O4 and Fe3O4@NaGdF4:Yb/Er@NaGdF4:Yb/Er NPs are shown in Fig. 2B. The field-dependent magnetization plots illustrate that Fe3O4 NPs are superparamagnetic with saturation magnetization value of 20.5 emu g−1. While Fe3O4@NaGdF4:Yb/Er@NaGdF4:Yb/Er NPs have a saturation magnetization value of 1.1 emu g−1 (inset in Fig. 2B). The markedly reduced value should be caused by the thick shells and organic OA molecules coated on magnetic NPs.
The up-conversion emission spectra of Fe3O4@NaGdF4:Yb/Tm@NaGdF4:Yb/Tm, Fe3O4@NaGdF4:Yb/Ho@NaGdF4:Yb/Ho and their corresponding photographs under 980 NIR excitation are presented in Fig. 3. As shown, the Tm3+ and Ho3+ based samples exhibit bright blue and yellow–green emissions upon 980 nm irradiation. In Fig. 3A, two intense blue emissions at 450 and 475 nm and a weaker red emission at 652 nm can be ascribed to the 1D2 → 3F4, 1G4 → 3H6 and 1G4 → 3F4 transitions of Tm3+.20 In Fig. 3B for the Fe3O4@NaGdF4:Yb/Ho@NaGdF4:Yb/Ho NPs, the dominating green emission at 541 nm can be associated with the 5F4, 5S2 levels to the 5I8 ground state. The weak blue emission at 475 nm originating from 5F3 → 5I8 and red emission at 645 nm from 5F5 → 5I8 transition are obvious.15
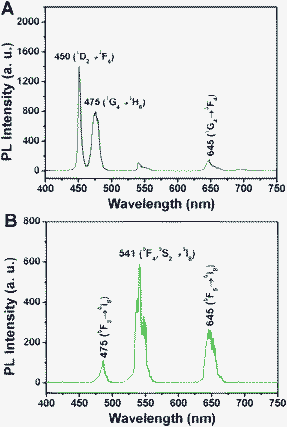 | ||
| Fig. 3 Up-conversion emission spectra of (A) Fe3O4@NaGdF4:Yb/Tm@NaGdF4:Yb/Tm, (B) Fe3O4@NaGdF4:Yb/Ho@NaGdF4:Yb/Ho and their corresponding photographs under 980 irradiation (insets). | ||
In conclusion, monodisperse, multicoloured and magnetic Fe3O4@NaGdF4:Ln@NaGdF4:Ln core–shell NPs have been successfully prepared via a seed-growth high-temperature thermolysis method. The as-prepared core–shell structured colloidal NPs show good monodispersity, uniform particle sizes. Notably, the core–shell structure markedly enhances the up-conversion emission intensity and also endows them with magnetic properties. These favorable features of the bifunctional material make it highly potential in biological field.
We are grateful to the National High Technology Research Program of China (2011AA03A407), National Basic Research Program of China (2010CB327704), National Natural Science Foundation of China (NSFC 20871035), Research Fund for the Doctoral Program of Higher Education of China (20112304110021) and the Fundamental Research Funds for the Central Universities of China HEUCF201210009).
References
- (a) Y. S. Liu, D. T. Tu, H. M. Zhu, R. F. Li, W. Q. Luo and X. Y. Chen, Adv. Mater., 2010, 22, 3266 CrossRef CAS; (b) F. Y. Liu, L. D. Carlos, R. A. S. Ferreira, J. Rocha, M. C. Ferro, A. Tourrette, F. Quignard and M. Robitzer, J. Phys. Chem. B, 2010, 114, 77 CrossRef CAS; (c) F. Caruso, A. S. Susha, M. Giersig and H. Möhwald, Adv. Mater., 1999, 11, 950 CrossRef CAS.
- (a) Y. H. Deng, W. L. Yang, C. C. Wang and S. K. Fu, Adv. Mater., 2003, 15, 1729 CrossRef CAS; (b) V. Salgueirinó-Maceira, M. A. Correa-Duarte, M. Spasova, L. M. Liz-Marzán and M. Farle, Adv. Mater., 2006, 16, 509 Search PubMed; (c) Y. Chan, J. P. Zimmer, M. Stroh, J. S. Steckel, R. K. Jain and M. G. Bawendi, Adv. Mater., 2004, 16, 2092 CrossRef CAS.
- (a) S. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989 CrossRef CAS; (b) S. L. Gai, P. P. Yang, C. X. Li, W. X. Wang, Y. L. Dai, N. Niu and J. Lin, Adv. Funct. Mater., 2010, 20, 1166 CrossRef CAS.
- T. Hyeon, Chem. Commun., 2003, 927 RSC.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706 CrossRef CAS.
- K. F. Schrum, J. M. Lancaster, S. E. Johnston and S. D. Gilman, Anal. Chem., 2000, 72, 4317 CrossRef CAS.
- X. Brokmann, J. P. Hermier, G. Messin, P. Desbiolles, J. P. Bouchaud and M. Dahan, Phys. Rev. Lett., 2003, 90 Search PubMed.
- H. T. Wong, H. L. W. Chan and J. H. Hao, Appl. Phys. Lett., 2009, 95, 022512 CrossRef.
- (a) K. S. Sohn, S. Lee, R. J. Xie and N. Hirosaki, Appl. Phys. Lett., 2009, 95, 121903 CrossRef; (b) C. C. Lin, Z. R. Xiao, G. Y. Guo, T. S. Chan and R. S. Liu, J. Am. Chem. Soc., 2010, 132, 3020 CrossRef CAS.
- L. Q. Xiong, T. S. Yang, Y. Yang, C. J. Xu and F. Y. Li, Biomaterials, 2010, 31, 7078 CrossRef CAS.
- (a) P. P. Yang, Z. W. Quan, C. X. Li, X. J. Kang, H. Z. Lian and J. Lin, Biomaterials, 2008, 29, 4341 CrossRef CAS; (b) L. Y. Wang and Y. D. Li, Chem. Commun., 2006, 24, 2557 RSC; (c) Q. Ju, D. T. Tu, Y. S. Liu, R. F. Li, H. M. Zhu, J. C. Chen, Z. Chen, M. D. Huang and X. Y. Chen, J. Am. Chem. Soc., 2012, 134, 1323 CrossRef CAS; (d) D. T. Tu, L. Q. Liu, Q. Ju, Y. S. Liu, H. M. Zhu, R. F. Li and X. Y. Chen, Angew. Chem., Int. Ed., 2011, 50, 6306 CrossRef CAS; (e) Q. Ju, Y. S. Liu, D. T. Tu, H. M. Zhu, R. F. Li and X. Y. Chen, Chem.–Eur. J., 2011, 17, 8549 CrossRef CAS; (f) Z. L. Wang, J. H. Hao, H. L. W. Chan, G. L. Law, W. T. Wong, K. L. Wong, M. B. Murphy, T. Su, Z. H. Zhang and S. Q. Zeng, Nanoscale, 2011, 3, 2175 RSC.
- J. Shen, L. D. Sun and C. H. Yan, Dalton Trans., 2008, 5687 RSC.
- (a) Z. L. Wang, Z. W. Quan, P. Y. Jia, C. K. Lin, Y. Luo, Y. Chen, J. Fang, W. Zhou, C. J. O. Connor and J. Lin, Chem. Mater., 2006, 18, 2030 CrossRef CAS; (b) C. R. De Silva, S. Smith, I. Shim, J. Pyun, T. Gutu, J. Jiao and Z. P. Zheng, J. Am. Chem. Soc., 2009, 131, 6336 CrossRef CAS.
- (a) M. M. Lezhnina, T. Jüstel, H. Kätker, D. U. Wiechert and U. H. Kynast, Adv. Funct. Mater., 2006, 16, 935 CrossRef CAS; (b) J. R. DiMaio, C. Sabatier, B. Kokuoz and J. Ballato, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1809 CrossRef CAS.
- (a) C. X. Li, X. M. Liu, P. P. Yang, C. M. Zhang, H. Z. Lian and J. Lin, J. Phys. Chem. C, 2008, 112, 2904 CrossRef CAS; (b) F. Vetrone, R. Naccache, V. Mahalingam, C. G. Morgan and J. A. Capobianco, Adv. Funct. Mater., 2009, 19, 2924 CrossRef CAS.
- (a) J. Park, K. An, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, N.-M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891 CrossRef CAS; (b) Z. Q. Li, Y. Zhang and S. Jiang, Adv. Mater., 2008, 20, 4765 CrossRef CAS.
- (a) Z. G. Chen, H. L. Chen, H. Hu, M. X. Yu, F. Y. Li, Q. Zhang, Z. G. Zhou, T. Yi and C. H. Huang, J. Am. Chem. Soc., 2008, 130, 3023 CrossRef CAS; (b) H. Hu, M. X. Yu, F. Y. Li, Z. G. Chen, X. Gao, L. Q. Xiong and C. H. Huang, Chem. Mater., 2008, 20, 7003 CrossRef CAS.
- X. M. Zhang, Z. W. Quan, J. Yang, P. P. Yang, H. Z. Lian and J. Lin, Nanotechnology, 2008, 19, 075603 CrossRef.
- (a) Z. L. Wang, J. H. Hao and H. L. W. Chan, J. Mater. Chem., 2010, 20, 3178 RSC; (b) F. Zhang and D. Y. Zhao, ACS Nano, 2009, 3, 159 CrossRef CAS; (c) F. Zhang, Y. Wan, Y. F. Shi, B. Tu and D. Y. Zhao, Chem. Mater., 2008, 20, 3778 CrossRef CAS; (d) J. H. Hao, Y. Zhang and X. H. Wei, Angew. Chem., Int. Ed., 2011, 50, 6876 CrossRef CAS; (e) F. Wang, R. R. Deng, J. Wang, Q. X. Wang, Y. Han, H. M. Zhu, X. Y. Chen and X. G. Liu, Nat. Mater., 2011, 10, 968 CrossRef CAS.
- (a) H. C. Lu, G. S. Yi, S. Y. Zhao, D. P. Chen and L. H. Guo, J. Mater. Chem., 2004, 14, 1336 RSC; (b) J. F. Suyver, J. Grimm, M. K. van Veen, D. Biner, K. W. Kramer and H. U. Gudel, J. Lumin., 2006, 117, 1 CrossRef CAS; (c) G. Z. Ren, S. J. Zen and J. H. Hao, J. Phys. Chem. C, 2011, 115, 20141 CrossRef CAS; (d) H. T. Wong, F. Vetrone, R. Naccache, H. L. W. Chan, J. H. Hao and J. A. Capobianco, J. Mater. Chem., 2011, 21, 16589 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental detail, XRD patterns, EDS spectra, FT-IR spectrum. See DOI: 10.1039/c2ra20070h |
| This journal is © The Royal Society of Chemistry 2012 |