Hydrogen storage and selective carbon dioxide capture in a new chromium(III)-based infinite coordination polymer†
Jian
Zhang
a,
Lixian
Sun
*ac,
Fen
Xu
*b,
Fen
Li
a,
Huai-Ying
Zhou
c,
Feng-Lei
Huang
d,
Zelimir
Gabelica
e and
Christoph
Schick
f
aMaterials and Thermochemistry Laboratory, Dalian Institute of Chemical Physics, 457 Zhongshan Road, Dalian, 116023, P. R. China. E-mail: lxsun@dicp.ac.cn; Fax: 86 411 8437 9213; Tel: 86 411 8437 9213
bInstitute of Chemistry for Functionalized Materials, College of Chemistry and Chemical Engineering, Liaoning Normal University, 850 Huanghe Road, Dalian, 116029, P. R. China. E-mail: xufen@lnnu.edu.cn;
cDepartment of Material Science & Engineering, Guilin University of Electrical Technology, Guilin, 541004, China
dState Key Laboratory of Explosion Science and Technology, Beijing Institute of Technology, Beijing, 100081, China
eUniversité de Haute Alsace, ENSCMu, Lab. LPI-GSEC, 3, Rue A. Werner, F-68094, Mulhouse Cedex, France
fInstitute of Physics, Universität Rostock, Rostock D-18059, Germany
First published on 13th February 2012
Abstract
A new chromium(III)-based infinite coordination polymer (ICP) was synthesized under solvothermal conditions. The morphology, particle size and the textural porosity of the resulting solids could be monitored and controlled by adjusting the volume ratio of the solvent mixture (1,4-dioxane/N,N′-dimethylformamide). Two differently textured samples were further compared using powder X-ray diffraction, scanning electron microscopy, FT-IR spectroscopy, solid-state UV-Vis spectroscopy and thermogravimetric analysis. Their sorption capacities for H2, CO2 and CH4 were measured by using volumetric gas adsorption, while the isosteric heats of adsorption for the same gasses were calculated based on gas sorption data at different temperatures. Both samples showed a remarkable selectivity for CO2 over CH4 at 273 K. H2 and CO2 sorption capacities, (respectively 16.9 mg g−1 at 77 K and 817 mm Hg and 168 mg g−1 at 273 K and 760 mm Hg) of the sample composed of the nanosized particles, are comparable or even superior to values reported for most MOFs and ZIFs under similar conditions.
Introduction
In the past decade, there have been numerous reports on the synthesis and characterization of metal–organic frameworks (MOFs) or coordination polymers (CPs). Such materials proved very useful in gas storage, separation, catalysis, and magnetism, especially in hydrogen storage and carbon dioxide capture.1 Recently, several groups have proposed a methodology to produce amorphous microparticles that readily assemble to form so-called infinite coordination polymers (ICPs), via solvent-induced precipitation, microemulsion, polymerization and solvothermal methods.2 ICPs are an attractive new class of materials currently used for innovative applications in drug delivery, imaging probes, photonics and biosensing.3 Gas sorption studies on ICP particles with permanent porosity in the dry state have been reported in only a few cases.4 In particular, Mirkin et al. claimed that a variety of such amorphous ICPs were capable of adsorbing different gaseous molecules and could interestingly be used for hydrogen storage.4f However it appears that the selective adsorption of CO2 over CH4 in porous materials, especially at ambient temperature and pressure, still remains challenging.5The present approach deals with ICP-type materials based on Cr(III) ions as the connecting metal. Cr(III) proved a good candidate to build flexible macro frameworks with organic ligands. Crystalline MOF's MIL-53 and the giant mesoporous structure MIL-101 are among the most famous examples of Cr(III)–benzene dicarboxylate (BDC) associations.6 Such materials were generated under hydrothermal conditions where the pH and amount of water were the main parameters to direct their specific structures.6c On the contrary, under (partial or total) solvothermal conditions, amorphous ICP-type assemblies are more readily generated using the same ingredients. In a recent report, a Cr(III)–BDC superstructure based on irregular packing of microsized amorphous nanospheres that exhibited a mixed (textural) micro- and mesoporosity, could be prepared in ambient conditions (room temperature) using non-aqueous solvents, namely a mixture of N,N′-dimethylformamide (DMF), trimethylamine and n-hexane.4e As this material showed significantly promising hydrogen uptake at high pressures, we wanted to explore in depth the possibility of synthesizing similar coordination polymers at high temperature (autoclave, autogenous pressure), through carefully selecting and varying the solvent system, with an aim of preparing materials with optimized morphologies (by controlling the particle size and their packing) and high surface areas required for an increased uptake of various gasses, more specifically of hydrogen.
Herein, we report original synthesis conditions leading to morphology-controlled infinite coordination polymer particles constructed using chromium(III) chloride hexahydrate and 1,4-benzenedicarboxylic acid (1,4-H2BDC) under solvothermal conditions. The use of pure DMF in which the ingredients are highly soluble was aimed at generating and stabilizing amorphous and defected particles, which are required for a high hydrogen uptake (see discussion). Hydrogen, carbon dioxide and methane sorption capacities of the differently textured materials were measured by using volumetric gas adsorption. The corresponding heats of adsorption were calculated based on gas adsorption data at different temperatures.
Experimental
All reagents were obtained from commercial sources at analytical grade and were used as received, unless otherwise noted.Synthesis of infinite coordination polymer particles
In a typical synthesis (sample 1), chromium(III) chloride hexahydrate (0.533 g, 2.0 mmol) and 1,4-H2BDC (0.332 g, 2.0 mmol) were dissolved in pure DMF solution (20 ml). The reactants were stirred before the clear solution was poured in a 30 ml Teflon-coated stainless steel autoclave. The autoclave was heated to 190 °C and maintained at that temperature for 3 days. After natural cooling to room temperature, the colloidal product obtained was filtered, washed thoroughly with DMF and finally dried in vacuum at 50 °C overnight to yield a dark green coarse powder. The as-synthesized sample was then heated at 200 °C for about 8 h in static air to remove the solvents (DMF and traces of water) impregnated on, or entrapped within the particles. The resulting dry solid was referred to as sample 1.The use of mixed solvents instead of pure DMF, thus involving 1,4-dioxane and DMF with the volume ratio (v/v, 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50), yielded a powder of olive-green color and was composed of 2 to 3 μm microparticles (sample 2).
:50), yielded a powder of olive-green color and was composed of 2 to 3 μm microparticles (sample 2).
Based on TGA and FT-IR data, samples 1 and 2 were deduced to have the same chemical formula: Cr(OH)·BDC.
Typically, in order to compare their different properties, samples 1 and 2 were prepared by hand milling and then shaped by a press under 2 MPa pressure. The pellets obtained were denoted as samples 1′ and 2′, respectively. All samples were heated at 200 °C for about 8 h in static air prior to characterization and testing, unless otherwise noted. All these infinite coordination polymer particles proved stable in air, water (moisture) and towards the other chemical agents used.
Low-pressure gas adsorption measurements
Nitrogen adsorption and desorption isotherms were measured at 77 K using an Autosorb-1 system after the samples were first degassed at 160 °C for 6 h. Specific surface areas (SSA) were determined by the BET method. The micropore surface area (Smicro.) was calculated by the t-plot method from an N2 adsorption–desorption isotherm, and the external surface area (Sext.) was evaluated by subtracting the t-plot micropore area from the BET specific surface area. The total pore volumes (Vt) and pore size distributions for all the coordination polymers were determined by analyzing the N2 isotherm at 77 K using non-local density functional theory (NLDFT) implementing a hybrid kernel based on a silica model containing cylindrical pores as implemented in the Autosorb-1 software package.7Low pressure hydrogen adsorption analyses were performed using the same Autosorb-1 system over a pressure range of 0–820 mm Hg at 77 K with the universal dewar filled only with liquid nitrogen. To obtain controllable temperatures such as 80 K, 85 K, and 90 K for calculating the heats of H2 adsorption, a customized Optistat®DN cryostat was used, which has been developed specially for Autosorb-1 analyzers by Quantachrome Instruments, in cooperation with OXFORD Instruments (Abingdon, U.K.). The cryostat option (interfaced Autosorb-1, ITC503 and the OptistatDN cryostat bath) allows one to run gas adsorption experiments over a wide range of controllable temperatures, from 77 K to 200 K, by using only liquid nitrogen as coolant.
Low pressure carbon dioxide and methane adsorption analyses were performed using the same Autosorb-1 system over a pressure range of 0–780 mm Hg at 273 K with an ice water bath and at room temperature (295 K for CO2 and 296 K for CH4).
Determination of isosteric heats of adsorption
The isosteric heats (Qst) of gas (H2, CO2 and CH4) adsorption on the coordination polymers were calculated as a function of surface coverage, by the Clausius–Clapeyron equation:8| Qst = −R × d(lnP)/d(1/T) | (1) |
High-pressure H2 adsorption measurements
High pressure hydrogen adsorption was performed using a gas reaction controller (GRC) from Advanced Materials Corporation (AMC) over a pressure range of 0–33 atm. Volumetric measurements at 77 K were carried out by using the liquid nitrogen cryostat system (STVP-200 continuous flow cryostat), for which the liquid level was maintained throughout the experiment. Before all measurements, the materials were activated in vacuum at 160 °C overnight using turbomolecular pump units to remove any physisorbed water or volatile impurities. Before hydrogen adsorption, blank tests were performed to ensure proper calibration and functioning of the instrumentation according to the operation manual of the gas reaction controller. Leak testing was also performed during each measurement by checking for soap bubbles at potential leak points and by observing the pressure changes of the corresponding pressure sensors. The skeletal densities were measured using a Micromeritics AccuPyc 1330 densimeter.Other physical measurements
Powder X-ray diffraction patterns (XRD) were collected with an X′Pert PRO X-ray diffractometer operating at 40 kV and 40 mA with Cu Kα radiation (λ = 1.5418 nm). FT-IR spectra were recorded on a Nicolet 380 infrared spectrometer using KBr pellets, in the 400–4000 cm−1 frequency range. Solid-state UV-Vis spectra were recorded on a Cintra 20 spectrometer. All scanning electron microscopy (SEM) images were obtained using a JSM-6360 LV SEM instrument operating at 20 kV. TG curves were recorded on a thermogravimetric analyzer (TherMax 500) at a heating rate of 10 °C min−1, in air flow.Results and discussion
Physico-chemical characterization of the various Cr(III)·BDC infinite coordination polymer samples
SEM pictures indicate that both the morphologies and the particle sizes of the two samples (sample 1 and sample 2) markedly differ. Fig. 1a shows that nanoparticles of about 100 nm are obtained when pure DMF solvent is used (sample 1). For a 1,4-dioxane/DMF volume ratio of 50:50 (sample 2), the quasi spherical particle size increases from nano-scale (Fig. 1a) to micro-scale (Fig. 1b). The marked change in morphology and particle size of the same coordination polymers could be attributed to a decrease of the admixed solvent polarity that then influences the polymer solubility in the solvent admixture. For a 1,4-dioxane/DMF volume ratio of 25:75 (SEM picture shown in Fig. S1†), the particle size of the product is intermediate between that of samples 1 and 2.
 | ||
| Fig. 1 SEM images of infinite coordination polymer particles. Volume ratios of 1,4-dioxane to DMF, a: 0:100 (sample 1); b: 50:50 (sample 2). | ||
It is generally acknowledged that, under hydrothermal or solvothermal conditions, small (nanometric) crystals are more readily obtained from clear solutions that favor a rapid homogeneous nucleation and thereby impede the crystal’s extensive growth.9a This was, for example, observed for zeolites and microporous aluminophosphates crystallized in water and in DMF, respectively, in closed systems (autoclave).9 Moreover, the relatively high temperature used in such systems (160–180 °C) accelerates the nucleation kinetics so that the numerous particles rapidly cease their further growth.9d In the present case, as both the Cr(III) salt and BDC are solubilized in pure DMF but less readily in 1,4-dioxane/DMF mixtures, the first system leads to homogeneous nucleation and yields the smallest particles.
Obviously the packing of such spheres will yield superstructured materials composed of spherical aggregates with a textural mesoporosity that depends on the size of the nano (or micro) spheres. The smaller the particle size, the smaller the mesopore diameter, which is fully confirmed by the N2 sorption–desorption data (see below).
Powder XRD patterns of 1 and 2 are very similar (Fig. S2†). The two weak and broad diffraction peaks recorded above 2θ angles of 15° indicate that both compounds are amorphous.
The formation of coordination polymers from metal ions and carboxylic ligands can be characterized by FT-IR spectra.2a,2g,10 FT-IR spectra (Fig. S3†) of 1 and 2 exhibit the typical vibrational bands in the region 1400–1700 cm−1 that characterize carboxylic functions. These results clearly show that the carboxylate groups are coordinated to Cr3+ ions, as shown by a shift of the asymmetric COO stretching frequency from around 1695 cm−1 for the protonated forms to around 1618 cm−1 for the deprotonated forms. The sharp absorption band located at 1400 cm−1 can be assigned to the symmetric COO stretching. The presence of three other sharp absorption bands (1618 cm−1, 1587 cm−1 and 1400 cm−1) of the carboxylate groups occurring in our chromium coordination polymers is in agreement with those recorded for MIL-53lt, Cr(OH)·BDC·H2O, where Cr(OH)2+ and (BDC)2− are interlinked through a bridging mode.6b Solid-state UV-Vis spectra (Fig. S4†) for both samples 1 and 2, display the same four wide adsorption peaks in the region from 200 to 800 nm, which is in line with a similar build up of a random framework made from infinite chains of Cr(III) octahedra linked through BDC dianions.
TGA studies (Fig. S5†) of 1 and 2, which were pre-activated at 200 °C in air, show weight losses of 70% and 69% (theoretical value: 67.4%), respectively, recorded from 250 °C to 550 °C at a heating rate of 10 °C min−1 in air. This indicates that the samples, devoid from any pre-sorbed solvent (DMF), are stable in air. The residual solid has been identified as crystallized chromium oxide Cr2O3 using XRD.
To acquire more information about the porous structure of these materials, N2 adsorption isotherms were recorded for all the infinite coordination polymer particles at liquid nitrogen temperature, using a commercial micropore gas analyzer (Autosorb-1). The corresponding isotherms are shown in Fig. 2.
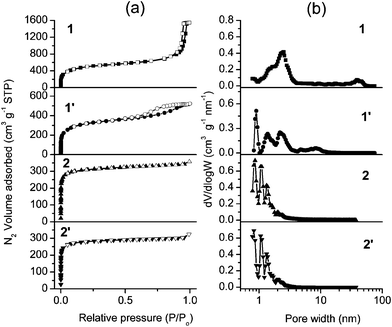 | ||
| Fig. 2 N2 adsorption–desorption isotherms (a) and pore size distributions (b) of coordination polymers calculated using NLDFT. In the isotherms, solid and open markers represent adsorption and desorption points respectively. | ||
The N2 adsorption–desorption isotherms of sample 1 showed a steep uptake below P/P0 = 0.02 due to the micropores filling, which is a characteristic of microporous solids.11 A hysteresis with a sharp reverse loop occurred in the high relative pressure region (Fig. 2a), which was associated with N2 capillary condensation in the interparticle void spaces generated by the stacking of the amorphous nanoparticles.11 This is in agreement with the above discussed SEM data. The BET specific surface area (SBET) of 1 is 1808 m2 g−1 (in Table 1), which is the highest surface area for an infinite coordination polymer reported so far.4Smicro. and Sext. of 1 extend up to 1345 m2 g−1 and 463 m2 g−1 respectively, these high values are characteristic of small nano-sized particles. Pore size distributions (PSDs) of 1, which were calculated by the nonlocal density functional theory (NLDFT) method (Fig. 2b),7 appeared very broad, extending from about 1 to 2 nm (micropores) and from 30 to 50 nm (mesopores). Samples 1 and 2 were shaped by a press under 2 MPa pressure. The pellets obtained were denoted as samples 1′ and 2′, respectively. Compared to 1, the total N2 volume adsorbed by 1′ decreased and the hysteresis loop broadened (Fig. 2a). The pore volume of 1′ is 0.77 cm3 g−1, which is far lower than the value obtained for 1 (2.33 cm3 g−1). SBET and Smicro. of 1′ are 1172 m2 g−1 and 898 m2 g−1, which are lower than those measured for 1 (in Table 1). The PSDs of 1′ are narrower than those of 1 and no pore size distribution was observed above 10 nm. The fact that both the mesopore and the micropore peak maxima had shifted to lower pore widths (respectively from 30–50 nm to less than 10 nm and from 1–2 nm to below 1 nm), indicates that the pore size and the specific surface area of 1 were markedly reduced by a press. The large interparticle nanopores (30–50 nm) completely disappeared by nanoparticle stacking under compression.
| CP | 1 | 1′ | 2 | 2′ |
|---|---|---|---|---|
| Pore volume/cm3 g−1 | 2.33 | 0.77 | 0.50 | 0.45 |
| BET specific surface area/m2 g−1 | 1808 | 1172 | 1210 | 1101 |
| Average pore width/nm | 2.50 | 0.90 | 0.86 | 0.82 |
| H2 uptake capacity/mmol g−1 | 8.47 | 5.29 | 8.20 | 7.52 |
| CO2 uptake capacity/mmol g−1 | 3.8 | 2.3 | 3.2 | 2.9 |
| CH4 uptake capacity/mmol g−1 | 0.7 | 0.6 | 0.9 | 0.7 |
The N2 adsorption–desorption isotherms of 2 and 2′ are of type I without a hysteresis loop (Fig. 2a),11 exhibiting a significant increase in the adsorbed volume at low relative pressures (P/P0 < 0.05), and reaching an almost horizontal plateau at higher pressures. This indicates that micro-sized particles of 2 and 2′ involving micropores do not aggregate to form a mesoporous structure (2–50 nm). SBET/Smicro. of 2 and 2′ now reach 1210/1143 m2 g−1 and 1101/1057 m2 g−1, while the pore volumes of 2 and 2′ reach 0.50 cm3 g−1 and 0.45 cm3 g−1 respectively. No notable changes in the N2 total volume adsorbed, nor in the PSD values of 2 and 2′ could be observed because of a less marked effect of the mechanical force on the porous structure of post-shaped microparticles (Fig. 2b).
The different types of isotherms of 1 and 2 confirm that their porous structures are totally different. In contrast to the solely microporous sample 2 that involves micro-sized particles, the product involving nano-sized particles (sample 1) is hierarchically a micro- and mesoporous infinite coordination polymer material.
Uptake capacities of H2, CO2 and CH4 for the various Cr(III)·BDC infinite coordination polymer samples
To investigate the influence of the pore size, the specific surface area and the chemical composition of the different infinite coordination polymer particles, the uptake capacities for H2 were measured using the same gas analyzer (Autosorb-1) at 77 K (Fig. 3). All samples display reversible H2 uptake, without hysteresis between the absorption and desorption isotherms, indicating that hydrogen is reversibly physisorbed.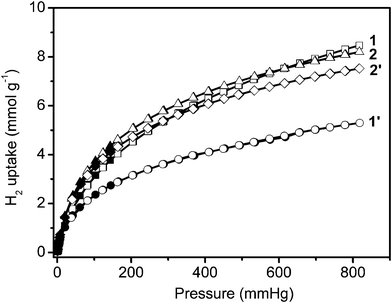 | ||
| Fig. 3 Low-pressure hydrogen adsorption isotherms at 77 K. In the isotherms, solid and open markers represent adsorption and desorption points respectively. | ||
The four infinite coordination polymer samples adsorb considerable amounts of H2. With increasing hydrogen pressure, the hydrogen uptake of 1 gradually exceeds that of 1′ (at 20 mm Hg), 2′ (at 330 mm Hg), and 2 (at 615 mm Hg) (Fig. 3). Finally, the hydrogen capacities of these samples increase in the order: 1′ (5.29 mmol g−1 or 10.6 mg g−1) < 2′ (7.52 mmol g−1 or 15.0 mg g−1) < 2 (8.20 mmol g−1 or 16.4 mg g−1) < 1 (8.47 mmol g−1 or 16.9 mg g−1) at 77 K and 817 mm Hg in Table 1. This also illustrates that the mesoporous sample 1 is logically the most sensitive to compression.
Finally, it is worth noting that the hydrogen uptake of 1 at 77 K and 817 mm Hg (16.9 mg g−1 or 1.45 wt%) reaches a higher value than those measured for most MOF type materials such as MOF-5 (1.32 wt%), MOF-177 (1.23 wt%) and for all ICPs reported in the recent literature.4,12
The high-pressure hydrogen storage performance was also measured with a commercial pressure–composition isotherm (PCI) unit provided by Advanced Materials Corporation (Fig. S6†). The hydrogen uptake capacities of the two samples are 25.5 mg g−1 for sample 2 and 36.3 mg g−1 for sample 1 at 77 K and 33 atm, values higher than those measured for ICPs and comparable to those recorded for HKUST-1 or MIL-53 (Al or Cr).4,13
We also determined the isosteric heat of adsorption for hydrogen in our coordination polymers as a function of H2 uptake, by comparing adsorption isotherms at 80 K, 85 K and 90 K obtained by using the AS1 cryostat option (Fig. S7†). The heat of H2 adsorption (Qst) was determined from the temperature dependence of the isotherms via the Clausius–Clapeyron equation.8 As shown in Fig. 4, the heat of adsorption lies in the ranges 6.7–5.4 kJ mol−1 for 1, 7.5–5.9 kJ mol−1 for 1′, 7.1–6.0 kJ mol−1 for 2, and 7.2–6.0 kJ mol−1 for 2′. The heat of adsorption of 1′ is greater than that of 1, which clearly indicates that the narrower pore size leads to a higher heat of adsorption. In the same way, the heats of adsorption of 2 and 2′ are larger than that of 1, due to their smaller PSDs. The heat of adsorption curves recorded for 2 and 2′ almost overlapped, due to their almost identical PSD and average pore size.
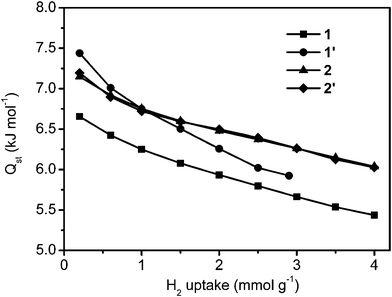 | ||
| Fig. 4 Heat of adsorption isotherms at different H2 loadings. | ||
Considering the hydrogen uptake capacity of these four samples at different hydrogen pressures, we clearly confirm that the effect of heat of adsorption on hydrogen uptake capacity is much larger than the effect of specific surface area at low pressure, whilst at high pressure, the effect is opposite.14
Furthermore, uptake capacities for CO2 and CH4 were measured at 273 K (Fig. 5 and 6). The four samples displayed a reversible CO2 and CH4 uptake, without hysteresis between the absorption and desorption isotherms, indicating that gases are reversibly physisorbed. In general, the trend of CO2 uptake is similar to what is observed for H2 uptake at 77 K. The CO2 uptake capacities of our 4 samples follow the same order: 1′ (52.6 cm3 g−1, 2.3 mmol g−1, 103 mg g−1) < 2′ (65.2 cm3 g−1, 2.9 mmol g−1, 128 mg g−1) < 2 (72.5 cm3 g−1, 3.2 mmol g−1, 143 mg g−1) < 1 (85.7 cm3 g−1, 3.8 mmol g−1,168 mg g−1) at 273 K and 760 mm Hg in Table 1. The CO2 uptake of 1 is the largest recorded for the four infinite coordination polymer samples prepared here, and is also higher compared to the corresponding values of some representative MOFs and ZIFs including MOF-5 (1.5 mmol g−1) and ZIF-100 (1.7 mmol g−1) at 273 K and low-pressure.15 However, the CH4 uptake capacities of these samples follow a different order: 1′ (12.6 cm3 g−1, 0.6 mmol g−1, 9.0 mg g−1) < 1 (16.2 cm3 g−1, 0.7 mmol g−1, 11.6 mg g−1) < 2′ (16.7 cm3 g−1, 0.7 mmol g−1, 11.9 mg g−1) < 2 (20.3 cm3 g−1, 0.9 mmol g−1, 14.5 mg g−1) at 273 K and 780 mm Hg (in Fig. 6 and Table 1).
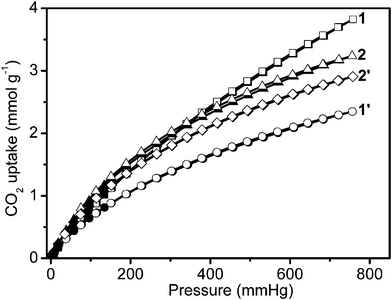 | ||
| Fig. 5 Low-pressure carbon dioxide adsorption isotherms at 273 K. In the isotherms, solid and open markers represent adsorption and desorption points respectively. | ||
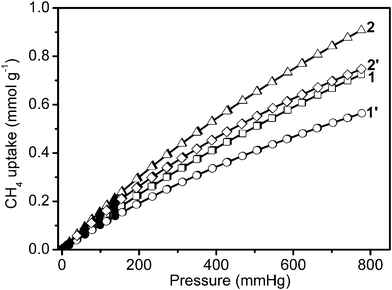 | ||
| Fig. 6 Low-pressure methane adsorption isotherms at 273 K. In the isotherms, solid and open markers represent adsorption and desorption points respectively. | ||
More interestingly, the four samples show similar adsorption trends (CO2 > CH4). We determined the isosteric heat of adsorption, by comparing adsorption isotherms of CO2 (at 273 K in Fig. 5 and at 295 K in Fig. S8†) and CH4 (at 273 K in Fig. 6 and at 296 K in Fig. S9†) in our coordination polymers as a function of gas uptake. Fig. 7 shows that CO2 has a higher isosteric heat of adsorption than CH4 (for CH4 = 18–21 kJ mol−1; and for CO2 = 29–38 kJ mol−1), which indicates the stronger affinity of CO2 for the material adsorption sites, as compared to CH4. It is possible that CO2 more readily binds to Cr(OH) sites through hydrogen-type bonding. Moreover, the significant difference in the adsorbed amounts of CO2 and CH4 could also be due to the fact that CO2 has a quadrupole moment, whereas CH4 is nonpolar. The effect of pore size on the heat of CO2 and CH4 adsorption is similar to what was observed for the H2 uptake at 77 K. The heat of CO2 and CH4 adsorption of sample 1 is lower than that of the other three samples at lower coverage.
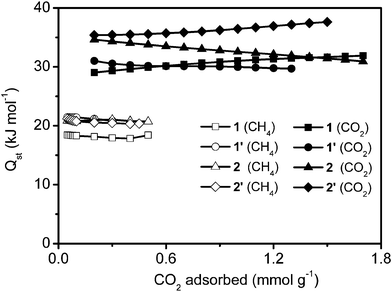 | ||
| Fig. 7 Heat of adsorption isotherms at different CO2 and CH4 loadings. | ||
The equilibrium molar loading ratios (α) of CO2/CH4 were measured at 273 K and 760 mm Hg and are expressed as α = (QCO2)/(QCH4), where (QCO2) and (QCH4) are the equilibrium uptakes of CO2 and CH4 at a given pressure taken from the corresponding pure-component isotherms. The equilibrium selectivities of the four samples studied in this work are shown in Fig. 8. The materials exhibit high selective adsorption of CO2 compared to CH4, especially at low pressure. The CO2 uptake for 1 is 8.6 times higher than the CH4 uptake at 4 mm Hg and 273 K, based on their single adsorption isotherms at 273 K (Fig. 5 and 6). The loading molar CO2/CH4 ratios decrease as expected with increasing pressure, especially in the pressure region below 200 mm Hg. When the pressure reaches 750 mm Hg, 1 still shows a selectivity of 5.4 times. This is much higher than for the other samples: 1′ (4.3), 2 (3.6) and 2′ (4.0), which were all measured under the same conditions. The CO2 uptake of the four samples is 3.6–5.4 times higher than the CH4 uptake. This markedly large and highly selective CO2 adsorption of sample 1 is an important property, indicating that the material can serve as an efficient CH4 purifier from CO2 at 273 K.
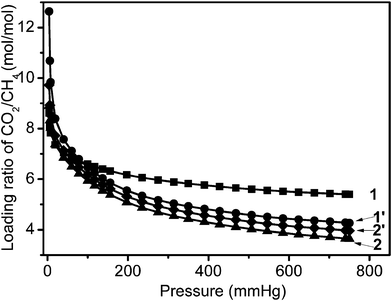 | ||
| Fig. 8 Loading molar ratios of CO2/CH4 calculated from the data shown in Fig. 5 and 6 up to pressures of 750 mm Hg and at 273 K. | ||
Conclusions
We have prepared a new Cr(III)-based amorphous infinite coordination polymer (ICP) interlinked with 1,4-dicarboxylate dianions, using a simple solvothermal method. The morphology and porous structure of this material could be monitored and controlled by adjusting the volume ratio of the solvent mixture (1,4-dioxane/DMF). Using solely DMF as the solvent resulted in the generation of small (nanosized) particles with a high surface area and a dual interparticle microporous and intraparticle (textural) mesoporous structure (sample 1). We clearly proved that the narrowing of the pore size results in a higher heat of adsorption for H2, CO2 and CH4 at low coverage. The four differently textured phases described here exhibit high surface area and high gas uptake capacities for H2 (at 77 K) and CO2 (273 K), but a low CH4 uptake (273 K). In particular, the H2 (at 77 K) and CO2 (at 273 K) uptake capacities of 1 are the largest measured for porous infinite coordination polymer particles and are comparable to other reported values for most MOFs and ZIFs measured under similar conditions. These unusually high uptakes were attributed to Cr(III) centers that were exposed when the weakly bound DMF was fully evacuated through the pre-heating of the as-synthesized Cr(OH)·BDC·DMF precursor, allowing hydrogen to bind at these incompletely coordinated sites.All samples showed a remarkable selectivity for CO2 over CH4 at 273 K. At low pressure, the CO2/CH4 selectivity is high (>8), indicating significant favorable interactions between CO2 and the material surface, possibly through H-bonding on Cr(OH) sites. However, this discrimination can also be attributed to the stronger interaction between the sample surface and CO2 that involves a quadrupolar moment. When the pressure is increased up to 750 mm Hg, 1 still shows a CO2/CH4 loading ratio of 5.4. It is concluded that sample 1 is a promising candidate for H2 storage (at 77 K), for CO2 capture, and as a CH4 purifier from CO2.
Acknowledgements
This work was financially supported by the “973 Project” (2010CB631303), NSFC (20833009, 20903095, 20873148, U0734005, 51071081, 51071146, 51101145 and 51102230), LNSFC (No. 20102224), Liaoning BaiQianWan Talents Program (No. 2010921050), Liaoning Education Committee (L2010223), the State Key Laboratory of Explosion Science and Technology, Beijing Institute of Technology (Grant No. KFJJ10-1Z) and IUPAC (Project No. 2008-006-3-100). The authors would like to thank Dr Zheling Zhang and Jeffrey Yang (Quantachrome Instruments, USA) for helpful discussions.References
- (a) N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127 CrossRef CAS; (b) M. Dinca, A. Dailly, Y. Liu, C. M. Brown, D. A. Neumann and J. R. Long, J. Am. Chem. Soc., 2006, 128, 16876 CrossRef CAS; (c) M. Latroche, S. Surblé, C. Serre, C. Mellot-Draznieks, P. L. Llewellyn, J. H. Lee, J. S. Chang, S. H. Jhung and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 8227 CrossRef CAS; (d) K. L. Mulfort, O. K. Farha, C. L. Stern, A. A. Sarjeant and J. T. Hupp, J. Am. Chem. Soc., 2009, 131, 3866 CrossRef CAS; (e) L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC; (f) H. J. Choi, M. Dinca, A. Dailly and J. R. Long, Energy Environ. Sci., 2010, 3, 117 RSC; (g) J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC; (h) M. Dinca and J. R. Long, Angew. Chem., Int. Ed., 2008, 47, 6766 CrossRef CAS.
- (a) M. Oh and C. A. Mirkin, Nature, 2005, 438, 651 CrossRef CAS; (b) X. P. Sun, S. J. Dong and E. K. Wang, J. Am. Chem. Soc., 2005, 127, 13102 CrossRef CAS; (c) W. J. Rieter, K. M. L. Taylor, H. Y. An, W. L. Lin and W. B. Lin, J. Am. Chem. Soc., 2006, 128, 9024 CrossRef CAS; (d) I. Imaz, D. Maspoch, C. Rodriguez-Blanco, J. M. Perez-Falcon, J. Campo and D. Ruiz-Molina, Angew. Chem., Int. Ed., 2008, 47, 1857 CrossRef CAS; (e) W. Lu, S. S. Y. Chui, K. M. Ng and C. M. Che, Angew. Chem., Int. Ed., 2008, 47, 4568 CrossRef CAS; (f) Z. Ni and R. I. Masel, J. Am. Chem. Soc., 2006, 128, 12394 CrossRef CAS; (g) S. Jung and M. Oh, Angew. Chem., Int. Ed., 2008, 47, 2049 CrossRef CAS.
- (a) A. M. Spokoyny, D. Kim, A. Sumrein and C. A. Mirkin, Chem. Soc. Rev., 2009, 38, 1218 RSC; (b) W. B. Lin, W. J. Rieter and K. M. L. Taylor, Angew. Chem., Int. Ed., 2009, 48, 650 CrossRef CAS.
- (a) Y. M. Jeon, G. S. Armatas, J. Heo, M. G. Kanatzidis and C. A. Mirkin, Adv. Mater., 2008, 20, 2105 CrossRef CAS; (b) W. Cho, H. J. Lee and M. Oh, J. Am. Chem. Soc., 2008, 130, 16943 CrossRef CAS; (c) Y. M. Jeon, G. S. Armatas, D. Kim, M. G. Kanatzidis and C. A. Mirkin, Small, 2009, 5, 46 CrossRef CAS; (d) H. J. Lee, W. Cho, S. Jung and M. Oh, Adv. Mater., 2009, 21, 674 CrossRef CAS; (e) Z. F. Xin, J. F. Bai, Y. M. Shen and Y. Pan, Cryst. Growth Des., 2010, 10, 2451 CrossRef CAS; (f) C. A. Mirkin, Y. M. Jeon and J. Heo, US Patent Appl. 12/336652, 2009 Search PubMed, assigned to Northwestern University.
- Q. Wang, J. Luo, Z. Zhong and A. Borgna, Energy Environ. Sci., 2011, 4, 42 RSC.
- (a) C. Serre, F. Millange, C. Thouvenot, M. Nogues, G. Marsolier, D. Louer and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519 CrossRef CAS; (b) G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040 CrossRef CAS; (c) N. A. Khan, J. W. Jun and S. H. Jhung, Eur. J. Inorg. Chem., 2010, 1043 CrossRef CAS.
- P. I. Ravikovitch, D. Wei, W. T. Chueh, G. L. Haller and A. V. Neimark, J. Phys. Chem. B, 1997, 101, 3671 CrossRef CAS.
- (a) F. Rouquerol, J. Rouquerol and K. Sing, Adsorption by Powders and Solids: Principles, Methodology, and Applications, Academic Press, 1999 Search PubMed; (b) S. Q. Ma, D. F. Sun, J. M. Simmons, C. D. Collier, D. Q. Yuan and H. C. Zhou, J. Am. Chem. Soc., 2008, 130, 1012 CrossRef CAS.
- (a) F. Gao and V. Valtchev, in Proc. XIVth Zeolite Forum, Kozierz, Poland, ed. V. Pashkova and M. Derewinski, 2007, 35 Search PubMed; (b) E. G. Derouane and Z. Gabelica, J. Solid State Chem., 1986, 64, 296 CrossRef CAS; (c) L. Vidal, J. L. Paillaud and Z. Gabelica, Microporous Mesoporous Mater., 1998, 24, 189 CrossRef CAS; (d) S. Mintova and V. Valtchev, Zeolites, 1993, 3, 136 Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds. Part B: Applications on Coordination, Organometallic and Bioinorganic Chemistry, Wiley, New-York, 2009, 424 Search PubMed.
- (a) K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS; (b) J. Rouquerol, D. Avnir, C. W. Fairbridge, D. H. Everett, J. H. Haynes, N. Pernicone, J. D. F. Ramsay, K. S. W. Sing and K. K. Unger, Pure Appl. Chem., 1994, 66, 1739 CrossRef CAS.
- J. L. C. Rowsell, A. R. Millward, K. S. Park and O. M. Yaghi, J. Am. Chem. Soc., 2004, 126, 5666 CrossRef CAS.
- (a) B. Panella, M. Hirscher, H. Putter and U. Muller, Adv. Funct. Mater., 2006, 16, 520 CrossRef CAS; (b) G. Férey, M. Latroche, C. Serre, F. Millange, T. Loiseau and A. Percheron-Guegan, Chem. Commun., 2003, 2976 RSC.
- M. Hirscher, B. Panella and B. Schmitz, Microporous Mesoporous Mater., 2010, 129, 335 CrossRef CAS.
- (a) H. Furukawa, J. Kim, N. W. Ockwig, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 11650 CrossRef CAS; (b) R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939 CrossRef CAS; (c) H. S. Choi and M. P. Suh, Angew. Chem., Int. Ed., 2009, 48, 6865 CrossRef CAS; (d) E. Neofotistou, C. D. Malliakas and P. N. Trikalitis, Chem.–Eur. J., 2009, 15, 4523 CrossRef CAS; (e) A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM, XRD, FT-IR, solid-state UV-Vis spectra, thermogravimetric analysis and gas sorption isotherms. See DOI: 10.1039/c2ra01188c/ |
| This journal is © The Royal Society of Chemistry 2012 |