Synthesis and immunostimulatory activity of diethanolamine-containing lipid A mimics†
Jordan D.
Lewicky
a,
Marina
Ulanova
b and
Zi-Hua
Jiang
*a
aDepartment of Chemistry, Lakehead University, 955 Oliver Road, Thunder Bay, Ontario, P7B 5E1, Canada. E-mail: zjiang@lakeheadu.ca; Fax: +1 807 346 7775; Tel: +1 807 766 7171
bMedical Sciences Division, Northern Ontario School of Medicine, Lakehead University, 955 Oliver Road, Thunder Bay, ON P7B 5E1, Canada.
First published on 6th January 2012
Abstract
Toll-like receptor (TLR) 4 plays important roles in the innate immunity and the development of adaptive immune responses. TLR4 ligands that can modulate the TLR4-mediated signalling pathways therefore have great potential for therapeutic applications. In this paper, we describe the synthesis of three lipid A mimics (2–4) as potential TLR4 ligands, in which a diethanolamine moiety is employed to replace the reducing end (D-glucosamine) of the archetypical lipid A disaccharide structure. Biological studies indicate that the lipid A mimic with six acyl chains (2) exhibits potent immune stimulatory activity in that it induces a significant increase in the ICAM-1 expression of human pre-monocytic THP-1 cells, as well as significant production of the cytokines TNF-α, IL-6, and IL-1β. The mimic with eight acyl chains (3) is inactive towards both the induction of ICAM-1 expression, and the cytokines TNF-α, and IL-6, yet induces significant production of IL-1β when tested at higher concentration. Finally, the lipid A mimic 4, a derivative of 2, that contains an additional 1-hydroxybutyl group as a result of an unexpected ring opening reaction of a tetrahydrofuran molecule, is active in all respects tested, albeit with reduced potency. These data suggest that diethanolamine-containing lipid A mimics can be potent immune stimulating agents.
1. Introduction
Lipopolysaccharide (LPS), a component of the outer membrane of Gram-negative bacteria, is perhaps the most effective of agents that induce the innate immune response.1 Structurally, LPS is primarily a large polysaccharide, however, the active principal of LPS responsible for the induction of the innate immune response lies in the terminal lipophilic moiety known as lipid A.2,3 The central structure of lipid A is a highly conserved β-(1→6) glycosidically linked di-D-glucosamine backbone bisphosphorylated at the 1-O- and 4′-O-position,4,5 as exemplified by the Escherichia coli lipid A structure (1, Fig. 1). LPS and lipid A trigger innate immune responses through the Toll-like receptor (TLR) 4/MD-2 complex, the activation of which leads to two distinct signalling pathways: a Myd88-dependent pathway and a TRIF-dependent pathway.6 The Myd88-dependent pathway ultimately results in the production of pro-inflammatory cytokines such as tumour necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6), whereas the TRIF-dependent pathway results in interferon-β (IFN-β) and nitric oxide production.6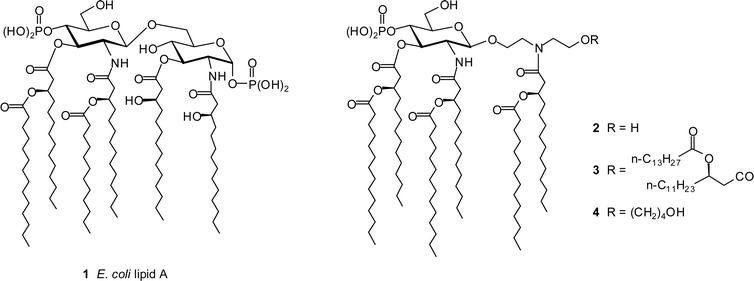 | ||
| Fig. 1 Structure of E. coli lipid A (1) and diethanolamine containing lipid A mimics 2–4. | ||
Recent advances in the understanding of the function of the TLR4/MD-2 complex have come in the form of protein crystal structures of the complex bound to both agonistic LPS, and antagonistic lipid IVa, the tetra-acyl biosynthetic precursor of E. coli lipid A.7,8 A large hydrophobic cavity is noted in MD-2, which is reported to contain all four lipid chains of lipid IVa, as compared to five of the six lipid chains of LPS. The remaining chain of LPS is exposed to the surface of MD-2, forming hydrophobic interactions with conserved phenylalanine residues in TLR4. These findings suggest that structural properties of MD-2 play critical roles in differentiating among varying lipid A structures as well as potentiating the biological responses.
As a result of its broad downstream implications, the TLR4 signalling pathway has attracted considerable attention in regards to potential immunotherapeutic manipulation, with efforts focused on both vaccine adjuvants and anti-sepsis treatments.9,10,11 Recent studies 12–17 with structurally defined synthetic lipid As and analogs have demonstrated that structural features such as the presence of a 3-deoxy-D-manno-octulosonic moiety, phosphorylation pattern, acylation pattern, the number of lipids, and lipid length are important for pro-inflammatory responses. For instance, the monophosphoryl lipid A (MPLA) derivatives are known to have reduced toxicity but retain potent immunostimulating activity. Studies have concluded that MPLA is a TLR4 agonist for the TRIF-dependent pathway. It seems that the removal of the anomeric phosphate group enables the molecule to affect the downstream signalling in a very subtle way. It has been suggested that the lack of pro-inflammatory activity of MPLA may be due to its ability to stimulate higher levels of the anti-inflammatory cytokine IL-10,49 or due to its inability to activate caspase-1 which is involved in the maturation of several proinflammatory mediators such as IL-1β and IL-18.50 Boons et al. have also shown that the potencies and efficacies of cytokine production are determined by transcriptional and post-transcription control mechanisms in cell-specific manners.14 A consequence of the potential toxicity of the TLR4-mediated inflammatory response has been a great deal of research focused on structure–activity relationships, aiming to separate the beneficial immunostimulatory activity from those endotoxic properties. Simplified lipid A structures with various acyclic molecular frameworks to mimic one or both D-glucosamine residues of the lipid A disaccharide backbone have been reported to display interesting immunostimulating activity.18–24,38 In those lipid A mimics with the reducing glucosamine residue replaced by an acyclic acylated aglycone that show potent immunostimulating activity, it appears that the location of the fatty acyl chain on the aglycone needs to be separated by a C2 or C3 linker from the glycosyl residue.20,23,38 Structure–activity relationship studies have also indicated that the position of the distal (reducing sugar) phosphate group is not strictly required.16
As part of our continuing work on the design and synthesis of TLR4 ligands as immunomodulating agents,25–28 we describe here new lipid A mimics (2–4, Fig. 1) wherein the reducing glucosamine residue of the disaccharide scaffold has been replaced by an acylated diethanolamine moiety. We have chosen diethanolamine as the acyclic aglycone to replace the reducing D-glucosamine based on the following consideration: (a) conservation of essential functional groups involved in TLR4/MD-2 ligand binding, namely, the phosphate and fatty acid chains; (b) conservation of the glycosidic linkage; (c) the appropriate location of each functional group in diethanolamine. Compounds 2–4 are monophosphorylated lipid A mimics with different numbers of fatty acyl chains. Compound 2 carries six fatty acyl chains while 3, with eight acyl chains, has been designed to examine the effect of lipid content on activity and is, to the best of our knowledge, the first example of a lipid A analog, either natural or synthetic, with more than seven fatty acyl chains. Compound 4 contains an additional 1-hydroxybutyl group, and is a side product encountered during our synthesis. Herein we report the synthesis and the preliminary biological activity of lipid A mimics 2–4.
2. Results and discussion
2.1 Synthesis
The synthesis of the designed lipid A mimics 2 and 3 began with the installation of lipid chains onto the diethanolamine acyclic scaffold (Scheme 1). As such, the amine moiety in diethanolamine was selectively acylated with dilipid acid 529 under the promotion of peptide coupling reagent O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluorophosphate (HBTU) to form glycosylation acceptor 6 in 68% yield. NMR spectral data indicated that the two ethanol residues in 6 were not identical, a consequence of the prohibited free rotation of the amide bond. The trimethylsilyl trifluoromethanesulfonate (TMSOTf) catalyzed glycosylation of 6 with known imidate donor 720 yielded the desired mono-glycosylation product 8 in 53% yield, along with the di-glycosylation product 9, which was isolated in 15% yield. Compound 8 existed as a mixture of two rotational isomers in an approximate ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 as a result of the presence of the secondary amide moiety. The desired β-glycosidic linkage in 8 was confirmed by its NMR spectral data (1H NMR: δ 4.78, d, J 8.0 Hz, H-1 from one isomer; and 13C NMR: δ 100.44 and 100.87, C-1 of two isomers).
:2 as a result of the presence of the secondary amide moiety. The desired β-glycosidic linkage in 8 was confirmed by its NMR spectral data (1H NMR: δ 4.78, d, J 8.0 Hz, H-1 from one isomer; and 13C NMR: δ 100.44 and 100.87, C-1 of two isomers).
 | ||
| Scheme 1 Reagents and conditions: (a) HBTU, (iPr)2NEt, DMF, 68%; (b) TMSOTf, CH2Cl2, 53% for 8 and 15% for 9. | ||
Removal of the N-Troc protecting group in 8via treatment with zinc in acetic acid–THF (1:4) provided free amine 10 (Scheme 2), which was found to be slowly converted to a new product during the reaction. This unexpected product was isolated and its MS data showed a molecular weight with an additional 72 mass units which corresponded to the incorporation of a THF molecule. Careful analysis of the respective NMR data led to the establishment of its structure as 11. THF ring opening is usually initiated by a strong Lewis acid 30,31 or an electrophilic reagent.32,33 With the present weakly acidic reaction condition, the ring opening of THF was suspected to be initiated by an electrophilic intermediate present in this reaction. Indeed, earlier studies showed that β-hydroxy alkylamides reacted with carboxylic acids via oxazolinium cation intermediates to form esters.34,35 Accordingly, we proposed a mechanism to account for the formation of product 11 (Scheme 3). Under acid catalysis, compound 10 could dehydrate to form oxazolinium cation II, which thereby reacted readily with nucleophiles present in the reaction mixture. Thus, the reaction of II with THF led to intermediate III, which was followed by a nucleophilic attack by water generated compound 11. Water was certainly present in the reaction mixture as the solvents were not dried. Accordingly, direct attack of intermediate II by water would regenerate compound 10. Acetic acid was likely the weakest nucleophile present, and thus no product carrying an additional acetate functionality was observed. Supporting this proposed mechanism was the clean conversion of compound 8 into desired amine 10 when the reaction was repeated with acetic acid as the sole solvent.
 | ||
| Scheme 2 Reagents and conditions: (a) Zinc dust, THF–HOAc (4:1), 25% for 10 and 55% for 11; (b) Zinc dust, HOAc, 85% for 10; (c) DCC, CH2Cl2, 65% for 12 and 16% for 13; (d) H2, Pd/C, THF–HOAc (4:1), 44% for 2 and 21% for 4; (e) H2, Pd/C, THF, 76% for 2 and < 5% for 4; (f) H2, Pd/C, THF, 85% for 3. | ||
 | ||
| Scheme 3 Postulated mechanism for the incorporation of a ring-opened THF residue to form product 11. | ||
The N, N′-dicyclohexylcarbodiimide (DCC) promoted coupling of amine 10 with dilipid acid 5 provided both hexa-acylated 12 and octa-acylated 13 in 65% and 16% yield, respectively. Acylation of a hydroxyl group normally does not occur under a DCC-promoted peptide coupling condition. Previous reports have shown that the hydroxyl group in β-hydroxy alkylamides displays higher reactivity than normal alcohols, the likes of which was noted to likely be the result of an intra-molecular hydrogen bond.36,37 Therefore, the formation of 13 might be due to an increased nucleophilicity of the hydroxyl group in 12 as a result of the presence of the same type of intra-molecular hydrogen bonding (Fig. 2). Through said hydrogen bonding, resonance structure 12-B is further stabilized and the electron density on the oxygen atom of the hydroxyl group is increased. Since enough material of 13 was obtained from this reaction, no attempt was made to prepare 13 separately through further acylation of 12 under typical hydroxyl acylation conditions.
 | ||
| Fig. 2 Intra-molecular hydrogen bonding in 12via a seven-member ring. | ||
Global de-benzylation of 12 was furnished via catalytic hydrogenation under atmospheric pressure in the presence of palladium on charcoal. Initially, the reaction was carried out in an acetic acid–THF (1:4) solvent system to give the desired product 2 in 44% yield, together with a side product 4 in 21% yield. The structures of 2 and 4 were confirmed by their 1H NMR and ESI-MS data. Side product 4 contained an additional 1-hydroxybutyl group reminiscent to that of compound 11 formed under similar reaction condition (acetic acid–THF, Scheme 2). Thus, the formation of 4 from compound 2 (and other partially debenzylated precursors) was most likely through the same mechanism described for the formation of 11 (Scheme 3). We also noticed that higher temperature (even at 30 °C) could significantly increase the conversion rate of compound 2 to 4. Acetic acid–THF mixture is a common solvent mixture used for the hydrogenation reaction of this type of compounds.25–27,38 We initially tried this reaction with this solvent mixture because we had no concept, at the time, about the mechanistic details for the formation of side product 11 aforementioned. Since the formation of the oxazolinium cation was acid catalysis dependent, we repeated this hydrogenation reaction using THF alone as the solvent. Indeed, this side reaction could be largely suppressed without acetic acid as the co-solvent: the side product 4 was formed in less than 5% and the yield of 2 was raised to 76%. Similar global debenzylation of 13 in THF provided 3 in 85% yield and its structure was confirmed by 1H NMR and high resolution ESI-MS data.
2.2 Biological evaluation
The activation of TLR4 by specific ligands leads to the release of pro-inflammatory cytokines and cellular adhesion molecules. The Human pre-monocytic THP-1 cell line expresses TLR4 as well as other receptors,39 and LPS has been shown to induce the expression of intercellular adhesion molecule-1 (ICAM-1) in these cells.40,41 Our initial studies of the immunostimulatory activity of lipid A mimics 2–4 involved the evaluation of these molecules in affecting the expression level of ICAM-1 by pre-monocytic THP-1 cells.Preliminary data indicates that compound 2 significantly increases the level of ICAM-1 expression in Human pre-monocytic THP-1 cells (Fig. 3). Compound 4 also exhibits significant activity, although it is less potent than 2. A maximum ICAM-1 expression level is achieved at a concentration of 2.0 μM for both 2 and 4, upon which further increases in their concentrations result in decreases in ICAM-1 expression levels. In contrast, no significant increase in ICAM-1 expression level is noted for the octa-acylated analog 3 at the highest concentration tested (4.0 μM). Importantly, all three compounds (2–4) show no detrimental effect on cell viability at the highest concentrations tested for each, as measured through the assay of cellular Trypan Blue exclusion.
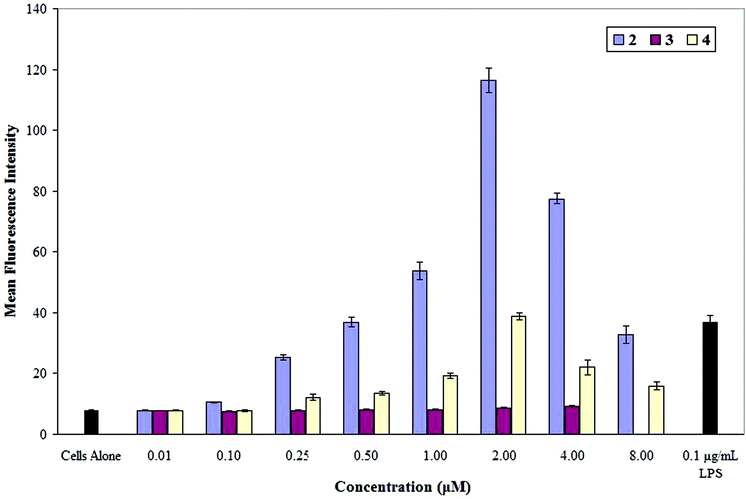 | ||
| Fig. 3 ICAM-1 expression by THP-1 cells after exposure to LPS and lipid A mimics (2–4). THP-1 cells were incubated for 18 h with increasing concentrations of 2–4. The resulting ICAM-1 expression was measured via immunostaining and flow cytometry analysis. The results are expressed as the mean fluorescence intensity and shown as the average of three separate experiments. | ||
ICAM-1 expression is NF-κB-dependent and induction of high levels of ICAM-1 occurs in response to various inflammatory mediators, including bacterial LPS, and pro-inflammatory cytokines, such as tumour necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and γ-interferon (γ-IFN).42,43 In an effort to further characterize the immunostimulatory properties of 2–4, the direct effects of the analogs on TNF-α, IL-6, and IL-1β cytokine production were measured. The pre-monocytic THP-1 cell line is weakly responsive in terms of cytokine production to immunostimulatory signals such as LPS.44 As such, terminal differentiation of the pre-monocytes was induced via 5 ng mL−1 of phorbol 12-myristate 13-acetate (PMA), the concentration of which was chosen to ensure that residual cytokine expression levels would be minimal and that small responses to weak stimuli were measurable.45
In general, the responses measured for all three cytokines mirror that of the ICAM-1 expression response, with compound 2 showing the greatest potency, compound 4 showing a slightly decreased potency, and compound 3 inducing very little to no detectable response (Fig. 4). Both 2 and 4 induce the highest level of TNF-α, IL-6 and IL-1β at the 9 μM maximum concentration tested, which is in contrast to the induction of ICAM-1 expression maximized at the concentration of 2 μM of the stimulus. The IL-1β level induced by 2 and 4 at 9 μM is 3–4 fold higher than that induced by E. coli LPS at the concentration of 0.01 μg mL−1. Interestingly, compound 3 at the 9 μM maximum concentration tested also induces significant IL-1β secretion while at the lower concentrations (up to 3 μM) the level of IL-1β is hardly detectable. IL-1β contributes to host defense against infection by augmenting the antimicrobial properties of phagocytes and initiating Th1 and Th17 adaptive immune responses.46 Production of IL-1β involves the proteolytic cleavage of pro-IL-1β by intracellular cysteine protease caspase-1,47 which is regulated by protein complexes called inflammasomes.48 Since 3 does not show any activity in the induction of ICAM-1 expression and other pro-inflammatory cytokines including TNF-α and IL-6, we believe that the induction of IL-1β by 3 may involve a different mechanism from that of analog 2 and 4.
 | ||
| Fig. 4 Cytokine production by differentiated THP-1 monocytes after stimulation with LPS and lipid A mimics (2–4). THP-1 monoyctes were incubated for 24 h with increasing concentrations 2–4. TNF-α (A), IL-6 (B), and IL-1β (C) in cell supernatants were measured via ELISAs. The results are shown as the average of two separate experiments. | ||
Structurally speaking, the hexa-acylated analog 2 shows the highest potency in the induction of ICAM-1 expression and pro-inflammatory cytokines (TNF-α, IL-6 and IL-1β). This immunostimulatory activity is retained with the additional 1-hydroxybutyl group incorporated in the diethanolamine moiety in 4, albeit with reduced potency. However, the increased number of lipid chains can have a more profound effect on the activity, as octa-acylated mimic 3 appears inactive in terms of inducing ICAM-1 expression, as well as inducing TNF-α and IL-6 production up to the highest concentration tested in each assay. Although significant IL-1β production is observed at the highest concentration of 3 tested, this may involve a different mechanism from TLR4/MD-2 activation which is presumably the mechanism of action of analog 2 and 4. Further studies are necessary to determine the precise mechanism through which these compounds exhibit their immunostimulatory activity, and ultimately, their potential applications.
In conclusion, we have successfully synthesized three monophosphorylated lipid A mimics 2–4 containing a diethanolamine moiety to replace the reducing D-glucosamine residue of the lipid A disaccharide structure. During our synthesis, we observed an interesting side reaction involving the glycosylated (N,N-diethanol)alkylamide residue. Under weak acid catalysis, the solvent THF was ring opened and attached to the (N,N-diethanol)alkylamide moiety, which was possibly initiated by an oxazolinium cation intermediate. From the biological studies, the hexa-acylated mimic 2 shows potent activity in inducing ICAM-1 expression by pre-monocytic THP-1 cells, as well as inducing TNF-α, IL-6, and IL-1β cytokine production in differentiated THP-1 monocytes, while derivative 4, which contains an additional 1-hydroxybutyl group, is less potent than 2. The octa-acylated analog 3 is inactive in the induction of ICAM-1 and other cytokines tested; however, it induces significant level of IL-1β at the concentration of 9 μM. Taken together, the results for derivatives 2–4 show that diethanolamine-containing lipid A mimics can be potent immunostimulating agents.
3. Experimental
3.1 General methods
All air and moisture sensitive reactions were performed under nitrogen atmosphere. All commercial reagents were used as supplied. Anhydrous dichloromethane was distilled over calcium hydride, whereas anhydrous N,N-dimethylformamide (DMF) was purchased from Aldrich. ACS grade solvents were purchased from Fisher Scientific and used for chromatography without distillation. TLC plates (silica gel 60 F254, thickness 0.25 mm) and silica gel 60 (40–63 μm) for flash column chromatography were purchased from SILICYCLE INC., Canada. 1H and 13C NMR spectra were recorded on a Varian Unity Inova 500 MHz spectrometer. Tetramethylsilane (TMS, δ 0.00 ppm) or solvent peaks were used as internal standards for 1H and 13C NMR spectra. The chemical shifts were given in ppm and coupling constants in Hz indicated to a resolution of ± 0.5 Hz. Multiplicity of proton signals is indicated as follows: s (singlet), d (doublet), dd (double doublet), t (triplet), q (quartet), m (multiplet), br (broad). Structural assignments were made using standard gCOSY and gHSQC methodology. NMR peaks belonging to primary lipid chains are denoted with an L subscript, whereas those belonging to secondary lipid chains are denoted with an L′ subscript. ESI mass spectra were measured on the Applied Biosystems Mariner Bio-Spectrometry Workstation at the University of Alberta, Canada. Optical rotations were measured with Perkin Elmer 343 Polarimeter at 22 °C.3.2 N,N-Bis(2-hydroxyethyl)-(R)-3-tetradecanoyloxytetradecanamide (6)
To a solution of diethanolamine (73 mg, 0.665 mmol) in DMF (3 mL), dilipid acid 529 (302 mg, 0.665 mmol), HBTU (262 mg, 0.698 mmol), and diisopropylethylamine (DIPEA, 0.24 mL, 1.40 mmol) were added. The mixture was stirred at room temperature for 16 h. The mixture was then concentrated, dissolved in water (30 mL), and extracted with EtOAc (3 × 30 mL). The combined organic layers were dried over Na2SO4, concentrated, and purified by flash column chromatography (hexane/EtOAc/MeOH, 1:1:0.125) to afford pure 6 (242 mg, 68%) as white solid. Rf 0.28 (hexane/EtOAc/MeOH, 1:1:0.1); [α]22D −3.7 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.84 (t, 6H, J 7.0 Hz, 2 × CH3), 1.16–1.33 (br m, 38H, 19 × CH2 of lipid), 1.51–1.62 (m, 4H, H–4L, H–3L′), 2.23 (t, 2H, J 7.5 Hz, H–2L′), 2.56 (dd, 1H, J 15.0, 7.5 Hz, H–2La), 2.68 (dd, J 15.0, 7.5 Hz, H–2Lb), 3.39–3.57 (m, 4H, HOCH2CH2NCH2CH2OH), 3.72–3.78 (m, 4H, HOCH2CH2NCH2CH2OH), 4.04–4.36 (br, 2H, OH × 2), 5.16–5.21 (m, 1H, H–3L); 13C NMR (125 MHz, CDCl3): δ 14.11 (CH3), 22.67 (CH2), 24.96 (CH2), 25.33 (CH2), 29.15 (CH2), 29.30 (CH2), 29.35 (CH2), 29.41 (CH2), 29.51 (CH2), 29.54 (CH2), 29.57 (CH2), 29.63 (CH2), 29.65 (CH2), 29.68 (CH2), 31.91 (CH2), 34.49 (C–2L′), 38.76 (C–2L), 50.29 & 52.39 (HOCH2CH2NCH2CH2OH), 60.44 & 60.74 (HOCH2CH2NCH2CH2OH), 71.40 (C–3L), 172.36 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 174.14 (CO); HRESI-MS (m/z) Calcd for C32H63NO5Na [M+Na]+: 564.4587, found: 564.4588.
O), 174.14 (CO); HRESI-MS (m/z) Calcd for C32H63NO5Na [M+Na]+: 564.4587, found: 564.4588.
3.3 N-(2-Hydroxyethyl)-N-{2-[6-O-benzyl-2-deoxy-4-O-(di-O-benzylphosphono)-3-O-((R)-3-tetradecanoyloxytetradecanoyl)-2-(2,2,2-trichloroethoxycarbonylamino)-β-D-glucopyranosyloxy]-ethyl}-(R)-3-tetradecanoyloxytetradecanamide (8) and N,N-bis{2-[6-O-benzyl-2-deoxy-4-O-(di-O-benzylphosphono)-3-O-((R)-3-tetradecanoyloxytetradecanoyl)-2-(2,2,2-trichloroethoxycarbonylamino)-β-D-glucopyranosyloxy]-ethyl}-(R)-3-tetradecanoyloxytetradecanamide (9)
A solution of 7 (460 mg, 0.36 mmol) and 6 (194 mg, 0.36 mmol) in dry CH2Cl2 (4 mL) in the presence of molecular sieves (4 Å, 2.0 g) was stirred under nitrogen for 30 min at room temperature. A solution of TMSOTf (0.01 M in dry CH2Cl2, 0.80 mL) was added drop wise in about 3 min. The mixture was stirred at room temperature for 1 h before a saturated sodium bicarbonate solution (10 mL) was added to quench the reaction. Solids were filtered out before the mixture was extracted with CH2Cl2 (3 × 25 mL). The combined organic phase was dried over Na2SO4, concentrated, and purified via repeated flash column chromatography (hexane/EtOAc, 2:1 and 3:4) to yield 8 (314 mg, 53%) and 9 (148 mg, 15%), both as colorless syrups.
For 8: Rf 0.40 (hexane/EtOAc, 3:4); [α]22D + 0.5 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.89 (t, 12H, J 7.0 Hz, 4 × CH3), 1.20–1.37 (br m, 76H, 38 × CH2 of lipid), 1.46–1.81 (br m, 8H H–4L ×2, H–3L′ × 2), 2.20–2.31 (m, 4H, H–2L′ × 2), 2.38–2.54 (m, 4, H–2L × 2), 3.25–3.40 (m, 1.4H, H–2 from one isomer, H–6b), 3.42–3.52 (m, 1.6H, H–2 from one isomer, H–6a), 3.56–3.84 (m, 8H, H-5, ROCH2CH2NCH2CH2OH, ROCH2CH2NCH2CH2OH), 4.03 (m, 2H, ROCH2CH2NCH2CH2OH), 4.38–4.57 (m, 3H, Troc–Ha, Troc–Hb, H–4), 4.64–4.73 (m, 2H, PhCH2), 4.78 (d, 0.4H, J 8.0 Hz, H–1 from one isomer), 4.85–4.92 (m, 4.6H, H–1 from one isomer, (PhCH2O)2P), 5.08–5.21 (m, 2H, H–3L × 2), 5.35 (dd, 0.4H, J 10.0, 10.0 Hz, H–3 from one isomer), 5.54 (dd, 0.6H, J 9.5, 9.5 Hz, H–3 from one isomer), 5.76 (d, 0.4H, J 8.0 Hz, NH from one isomer), 6.24 (d, 0.6H, J 7.0 Hz, NH from one isomer), 7.27–7.32 (m, 15H, Ar–H); 13C NMR (125 MHz, CDCl3): δ 14.34 (CH3), 22.90 (CH2), 25.19 (CH2), 25.23 (CH2), 25.30 (CH2), 25.56 (CH2), 25.77 (CH2), 29.38 (CH2), 29.42 (CH2), 29.53 (CH2), 29.57 (CH2), 29.66 (CH2), 29.75 (CH2), 29.78 (CH2), 29.79 (CH2), 29.82 (CH2), 29.87 (CH2), 29.90 (CH2), 32.13 (CH2), 34.26 (CH2), 34.51 (CH2), 34.62 (CH2), 34.64 (CH2), 34.74 (CH2), 34.79 (CH2), 34.84 (CH2), 38.70 (CH2), 39.38 (CH2), 39.61 (CH2), 47.84 (CH2N), 49.54 (CH2N), 50.84 (CH2N), 52.20 (CH2N), 56.75 (C–2), 60.35 (C–6 from one isomer), 61.60 (C–6 from one isomer), 68.12 (OCH2), 68.36 (OCH2), 68.64 (OCH2), 68.97 (OCH2), 69.85–69.97 (m, (PhCH2O)2P), 70.24 (C–3L), 70.27 (C–3L), 71.70 (C–3L), 71.91 (C–3 from one isomer), 72.52 (C–3 from one isomer), 72.15 (C–3L), 73.63 (Troc–CH2), 73.90 (C–4 from one isomer), 73.94 (C–5 from one isomer), 74.21 (C–5 from one isomer), 74.26 (d, J 5.0 Hz, C–4 from one isomer), 74.33 (d, J 5.0 Hz, C–4 from one isomer), 74.52 (PhCH2 from one isomer), 74.67 (PhCH2 from one isomer), 95.17 (Troc–CCl3), 100.44 (C–1 from one isomer), 100.87 (C–1 from one isomer), 127.64 (CH-Ar), 127.98 (CH-Ar), 128.08 (CH-Ar), 128.34 (CH-Ar), 128.56 (CH-Ar), 128.57 (CH-Ar), 128.62 (CH-Ar), 135.43 (d, J 2.5 Hz, C–Ar), 135.68 (d, J 2.5 Hz, C–Ar), 137.99 (C–Ar), 138.06 (C–Ar), 154.54 (CO Troc), 170.25 (CO), 170.50 (CO), 171.18 (CO), 171.95 (CO), 173.65 (CO) 173.80 (CO) 174.28 (CO); HRESI–MS (m/z) Calcd for C90H146Cl3N2O17P [M+Na]+: 1685.9282, found: 1685.9317.
For 9: Rf 0.36 (hexane/EtOAc, 2:1); [α]22D + 0.5 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.88 (t, 18H, J 7.0 Hz, 6 × CH3), 1.20–1.37 (br m, 114H, 57 × CH2 of lipid), 1.44–1.84 (br m, 12H H–4L × 3, H–3L′ × 3), 2.20–2.31 (m, 6H, H–2L′ × 3), 2.38–2.54 (m, 4H, H–2L × 2), 2.68–2.77 (m, 2H, H–2L), 3.26–3.43 (m, 3H, H–2 from one sugar, CH2N), 3.51–3.70 (m, 10H, H–2 from one sugar, H–5 from one sugar, H–6a from both sugars, H–6b from both sugars, CH2N, CH2O), 3.74–3.92 (m, 3H, H–5 from one sugar, CH2O), 4.36–4.54 (m, 6H, H–4 from both sugars, PhCH2 from both sugars), 4.58 (d, 1H, J 8.0 Hz, H–1 from one sugar), 4.63–4.80 (m, 5H, H–1 from one sugar, Troc–Ha from both sugars, Troc–Hb from both sugars), 4.86–4.93 (m, 8H, (PhCH2O)2P from both sugars), 5.08–5.16 (m, 3H, H–3L), 5.26 (dd, 1H, J 9.5, 9.5 Hz, H–3 from one sugar), 5.52 (dd, 1H, J 9.5, 9.5 Hz, H–3 from one sugar), 5.73 (d, 1H, J 8.0 Hz, NH from one sugar), 6.14 (d, 1H, J 8.0 Hz, NH from one sugar), 7.24–7.38 (m, 30H, Ar–H); 13C NMR (125 MHz, CDCl3): δ 14.17 (CH3), 22.73 (CH2), 25.01 (CH2), 25.04 (CH2), 25.09 (CH2), 25.13 (CH2), 25.69 (CH2), 29.19 (CH2), 29.28 (CH2), 29.36 (CH2), 29.40 (CH2), 29.42 (CH2), 29.49 (CH2), 29.57 (CH2), 29.62 (CH2), 29.64 (CH2), 29.69 (CH2), 29.71 (CH2), 29.74 (CH2), 31.96 (CH2), 33.97 (CH2), 34.28 (CH2), 34.43 (CH2), 34.47 (CH2), 34.72 (CH2), 38.30 (CH2), 38.93 (CH2), 39.20 (CH2), 45.01 (CH2N), 46.83 (CH2N), 46.87 (CH2N), 48.83 (CH2N), 56.39 (C–2 from one sugar), 56.50 (C–2 from one sugar), 60.21 (C–6 from one sugar), 61.47 (C–6 from one sugar), 67.94 (OCH2), 68.37 (OCH2), 68.41 (OCH2), 68.45 (OCH2), 69.64 (m, (PhCH2O)2P from both sugars), 69.97 (C–3L), 72.03 (C–3L), 72.07 (C–3L), 72.14 (C–3 from one sugar), 72. 44 (C–3 from one sugar), 73.38 (PhCH2 from one sugar), 73.42 (PhCH2 from one sugar), 73.78 (C–5 from one sugar), 73.84 (C–5 from one sugar), 73.96 (d, J 5.0 Hz, C–4 from one sugar), 74.05 (d, J 5.0 Hz, C–4 from one sugar), 74.35 (Troc–CH2 from one sugar), 74.61 (Troc–CH2 from one sugar), 95.51 (Troc–CCl3 from one sugar), 95.72 (Troc–CCl3 from one sugar), 100.57 (C–1 from one sugar), 102.03 (C–1 from one sugar), 127.61 (CH-Ar), 127.67 (CH-Ar), 128.01 (CH-Ar), 128.03 (CH-Ar), 128.11 (CH-Ar), 128.15 (CH-Ar), 128.36 (CH-Ar), 128.40 (CH-Ar), 128.58 (CH-Ar), 128.61 (CH-Ar), 128.65 (CH-Ar), 135.56 (m, C-Ar), 137.94 (C-Ar), 138.02 (C-Ar), 154.12 (CO Troc from one sugar), 154.51 (CO Troc from one sugar), 170.01 (CO), 170.46 (CO), 170.70 (CO), 173.28 (CO), 173.47 (CO), 174.11 (CO); HRESI–MS (m/z) Calcd for C148H229Cl6N3O29P2 [M+Na]+: 2807.3979, found: 2807.4086.
3.4 N-[2-(4-hydroxybutyloxy)-ethyl]-N-{2-[2-amino-6-O-benzyl-2-deoxy-4-O-(di-O-benzylphosphono)-3-O-((R)-3-tetradecanoyloxytetradecanoyl)-β-D-glucopyranosyloxy]-ethyl}-(R)-3-tetradecanoyloxytetradecanamide (11)
To a solution of 8 (100 mg, 0.06 mmol) in THF (4 mL) and glacial acetic acid (1 mL), zinc powder (500 mg) was added. The mixture was stirred at room temperature and the progress of the reaction monitored via TLC. After approximately 30 min, the complete consumption of 8 was noted. However, two different product spots were noted. Interestingly, the slow conversion of one of the product spots into the other was noted as the reaction was allowed to stir further. After stirring for approximately 4 h, the solid was filtered, washed with acetic acid (30 mL) and the filtrate concentrated in vacuo. The residue was dissolved in CH2Cl2 (100 mL) and washed with a saturated sodium bicarbonate solution (50 mL). The organic phase was dried with Na2SO4, concentrated, and the residue purified by flash column chromatography (hexane/EtOAc, 2:3) to obtain pure 11 (51 mg, 55%) as a colorless syrup. Rf 0.26 (4% MeOH in CH2Cl2); [α]22D + 0.2 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.89 (t, 12H, J 7.0 Hz, 4 × CH3), 1.11–1.40 (br m, 76H, 38 × CH2 of lipid), 1.41–1.74 (br m, 12H, H–4L × 3, H–3L′ × 3, ROCH2CH2CH2CH2OH), 2.18–2.31 (m, 4H, H–2L′ × 2), 2.38–2.45 (m, 1H, H–2L), 2.49–2.55 (m, 1.4H, H–2 from one isomer, H–2L), 2.60–2.78 (m, 4.6H, H–2 from one isomer, NH2, H–2L × 2), 3.33–3.39 (m, 1H, H–6a), 3.43–3.70 (m, 11H, H–5, H–6b, ROCH2CH2NCH2CH2OCH2CH2CH2CH2OH), 3.74–3.91 (m, 3H, ROCHHCH2NCH2CH2OCH2CH2CH2CH2OH), 4.01–4.06 (m, 1H, ROCHHCH2NCH2CH2OCH2CH2CH2CH2OH), 4.30 (d, 0.4H, J 8.0 Hz, H–1 from one isomer), 4.34–4.50 (m, 3.6 H, H–1 from one isomer, H–4, PhCH2), 4.85–4.94 (m, 4H, (PhCH2O)2P), 5.04–5.14 (m, 1.4H, H–3 from one isomer, H–3L), 5.19–5.24 (m, 1.6H, H–3 from one isomer, H–3L), 7.22–7.37 (m, 15H, Ar–H); 13C NMR (125 MHz, CDCl3): δ 22.92 (CH3), 22.73 (CH2), 25.12 (CH2), 25.17 (CH2), 25.21 (CH2), 25.24 (CH2), 25.36 (CH2), 25.62 (CH2), 25.67 (CH2), 27.76 (CH2), 28.26 (CH2), 29.36 (CH2), 29.41 (CH2), 29.42 (CH2), 29.44 (CH2), 29.53 (CH2), 29.55 (CH2), 29.59 (CH2), 29.63 (CH2), 29.67 (CH2), 29.75(CH2), 29.81 (CH2), 29.87 (CH2), 29.89(CH2), 29.91 (CH2), 29.93 (CH2), 31.31 (CH2), 31.89 (CH2), 31.96 (ROCH2CH2CH2CH2OH from one isomer), 32.01 (ROCH2CH2CH2CH2OH from one isomer), 32.07 (ROCH2CH2CH2CH2OH from one isomer), 32.14 (ROCH2CH2CH2CH2OH from one isomer), 34.56 (CH2), 34.60 (CH2), 34.77 (CH2), 34.82 (CH2), 38.79 (CH2), 39.23 (CH2), 40.01 (CH2), 40.06 (CH2), 45.69 (CH2N), 48.34 (CH2N), 49.44 (CH2N), 49.86 (CH2N), 51.33 (C–6 from one isomer), 52.69 (C–6 from one isomer), 60.03 (OCH2), 60.98 (OCH2), 61.40 (C–2 from one isomer), 62.24 (C–2 from one isomer), 62.43 (OCH2), 62.49 (OCH2), 67.95 (OCH2), 68.14 (OCH2), 68.56 (OCH2), 68.76 (OCH2), 69.91 (m, (PhCH2O)2P), 70.34 (C–3L), 70.38 (C–3 from one isomer), 71.33 (C–3 from one isomer), 71.52(C–3L), 73.58 (PhCH2 from one isomer), 73.62 (PhCH2 from one isomer), 73.94 (C–3 from one isomer), 74.06 (C–3 from one isomer), 74.20 (C–5 from one isomer), 74.25 (C–5 from one isomer), 74.59 (d, J 5.0 Hz, C–4 from one isomer), 74.66 (d, J 5.0 Hz, C–4 from one isomer), 100.04 (C–1 from one isomer), 104.06 (C–1 from one isomer), 127.83 (CH-Ar), 127.86 (CH-Ar), 128.22 (CH-Ar), 128.27 (CH-Ar), 128.32 (CH-Ar), 128.39 (CH-Ar), 128.56 (CH-Ar), 128.80 (CH-Ar), 128.85 (CH-Ar), 128.89 (CH-Ar), 135.70 (m, C-Ar), 138.12 (C-Ar), 170.83 (CO), 171.16 (CO), 171.29 (CO), 171.59 (CO), 173.68 (CO), 173.71 (CO), 173.75 (CO), 173.99 (CO); ESI–MS (m/z) Calcd for C91H154N2O16P [M+H]+: 1563.1, found: 1563.1.
3.5 N-(2-Hydroxyethyl)-N-{2-[6-O-benzyl-2-deoxy-4-O-(di-O-benzylphosphono)-3-O-((R)-3-tetradecanoyloxytetradecanoyl)-2-((R)-3-tetradecanoyloxytetradecanamido)-β-D-glucopyranosyloxy]-ethyl}-(R)-3-tetradecanoyloxytetradecanamide (12) and N-{2-[(R)-3-tetradecanoyloxytetradecanoyloxy]-ethyl)-N-{2-[6-O-benzyl-2-deoxy-4-O-(di-O-benzylphosphono)-3-O-((R)-3-tetradecanoyloxytetradecanoyl)-2-((R)-3-tetradecanoyloxytetradecanamido)-β-D-glucopyranosyloxy]-ethyl}-(R)-3-tetradecanoyloxytetradecanamide (13)
To a solution of 8 (320 mg, 0.19 mmol) in glacial acetic acid (5 mL), zinc powder (1.0 g) was added. The mixture was stirred at room temperature for 30 min and then filtered. The solid was washed with acetic acid (30 mL) and the filtrate concentrated in vacuo. The residue was dissolved in CH2Cl2 (150 mL) and washed with a saturated sodium bicarbonate solution (75 mL). The combined organic phase was dried with Na2SO4 and concentrated to obtain free amine 10 (244 mg, 85%) as a white solid.A mixture of amine 10 (244 mg, 0.16 mmol), 5 (160 mg, 0.33 mmol), and DCC (154 mg, 0.66 mmol) in dry CH2Cl2 (5 mL) was stirred at room temperature for 20 h. Water (0.50 mL) was added and the reaction mixture was stirred for a further 20 min. The solid was then filtered through a sintered glass funnel with a bed of Na2SO4. The filtrate was concentrated and the residue purified by repeated flash column chromatography (hexane/acetone, 5:1 and 4.5:1) to afford 12 (203 mg, 65%) and 13 (63 mg, 16%), both as colorless syrups.
For compound 12: Rf 0.31 (hexane/acetone, 4:1); [α]22D −2.2 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.89 (t, 18H, J 7.0 Hz, 6 × CH3), 1.21–1.39 (br m, 114H, 57 × CH2 of lipid), 1.48–1.72 (br m, 12H H–4L × 3, H–3L′ × 3), 2.18–2.56 (m, 10H, H–2L′ × 3, H–2L × 2), 2.76 (dd, 1H, J 15.5, 7.5 Hz, H–2La), 2.85 (dd. 1H, J 15.5, 5.0 Hz, H–2Lb), 3.18–3.23 (m, 0.6H, H–2 from one isomer), 3.29–3.37 (m, 2H, H–6a, H–6b), 3.44 (br s, 1H, OH), 3.52–3.86 (m, 7.4 H, H–2 from one isomer, H–5, ROCH2CH2NCH2CH2OH), 3.97– 4.08 (m, 2H, ROCH2CH2NCH2CH2OH), 4.41–4.52 (m, 3H, H–4, PhCH2), 4.63 (d, 0.4H, J 8.0 Hz, H–1 from one isomer), 4.82–4.94 (m, 4H, (PhCH2O)2P), 5.10 (m, 4H, H–1 from one isomer, H–3 from one isomer, C–3L × 3), 5.64 (dd, 0.6H, J 10.5, 8.5 Hz, H–3 from one isomer), 6.18 (d, 0.4H, J 8.0 Hz, NH from one isomer), 6.80 (d, 0.6H, J 8.0 Hz, NH from one isomer), 7.21–7.39 (m, 15H, Ar–H); 13C NMR (125 MHz, CDCl3): δ 14.15 (CH3), 22.73 (CH2), 25.05 (CH2), 25.17 (CH2), 25.20 (CH2), 25.31 (CH2), 25.38 (CH2), 25.69 (CH2), 29.21 (CH2), 29.25 (CH2), 29.26 (CH2), 29.28 (CH2), 29.32 (CH2), 29.36 (CH2), 29.39 (CH2), 29.42 (CH2), 29.45 (CH2), 29.50 (CH2), 29.52 (CH2), 29.56 (CH2), 29.62 (CH2), 29.64 (CH2), 29.66 (CH2), 29.71 (CH2), 29.73 (CH2), 29.74 (CH2), 29.76 (CH2), 29.80 (CH2), 31.96 (CH2), 31.98 (CH2), 34.19 (CH2), 34.41 (CH2), 34.47 (CH2), 34.53 (CH2), 34.60 (CH2), 34.66 (CH2), 38.24 (CH2), 38.60 (CH2), 39.25 (CH2), 39.46 (CH2), 40.85 (CH2), 41.51 (CH2), 47.60 (CH2N), 49.49 (CH2N), 51.21 (CH2N), 51.88 (CH2N), 54.77 (C–2 from one isomer), 56.40 (C–2 from one isomer), 60.15 (C–6 from one isomer), 60.97 (C–6 from isomer), 67.67 (OCH2), 68.25 (OCH2), 68.41 (OCH2), 68.66 (OCH2), 69.59 (m, (PhCH2O)2P), 70.14 (C–3L), 70.87 (C–3 from one isomer), 71.40 (C–3 from one isomer), 71.86 (C–3L), 72.15 (C–3L), 73.37 (PhCH2 from one isomer), 73.45 (PhCH2 from one isomer), 73.86 (C–5 from one isomer), 73.90 (C–5 from one isomer), 74.13 (d, J 5.0 Hz, C–4 from one isomer), 74.33 (d, J 5.0 Hz, C–4 from one isomer), 99.76 (C–1 from one isomer), 100.92 (C–1 from one isomer), 127.61 (CH-Ar), 127.66 (CH-Ar), 127.98 (CH-Ar), 128.00 (CH-Ar), 128.09 (CH-Ar), 128.11 (CH-Ar), 128.30 (CH-Ar), 128.35 (CH-Ar), 128.52 (CH-Ar), 128.54 (CH-Ar), 128.56 (CH-Ar), 128.58 (CH-Ar), 128.61 (CH-Ar), 128.67 (CH-Ar), 135.53 (m, C-Ar), 137.84 (C-Ar), 137.97 (C-Ar), 169.80 (CO), 170.52 (CO), 170.62 (CO), 170.77 (CO), 171.37 (CO), 171.69 (CO), 173.20 (CO), 173.33 (CO), 173.52 (CO), 173.56 (CO), 173.58 (CO), 174.37 (CO); HRESI–MS (m/z) Calcd for C115H197N2O18P [M+Na]+: 1948.4144, found: 1948.4183.
For compound 13: Rf 0.41 (hexane/acetone, 4:1); [α]22D −1.1 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.83 (t, 24H, J 7.0 Hz, 8 × CH3), 1.21–1.39 (br m, 152H, 76 × CH2 of lipid), 1.46–1.79 (br m, 16H, H–4L × 4, H–3L′ × 4), 2.17–2.58 (m, 14H, H–2L′ × 4, H–2L × 3), 2.73 (dd, 1H, J 16.0, 6.0 Hz, H–2La), 2.85 (dd, 1H, J 16.0, 5.0 Hz, H–2Lb), 3.12–3.17 (m, 0.6H, H–2 from one isomer), 3.23–3.41 (m, 2H, H–6a, H–6b), 3.49–3.78 (m, 6.4H, H–2 from one isomer, H–5, R1OCH2CH2NCH2CH2OR2, OCHH), 3.89–3.95 (m, 1H, OCHH), 4.11–4.17 (m, 2H, OCH2), 4.39–4.51 (m, 3H, H–4, PhCH2), 4.69 (d, 0.4H, J 8.5 Hz, H–1 from one isomer), 4.85–4.91 (m, 4H, (PhCH2O)2P), 5.06–5.24 (m, 4.6 H, H–1 from one isomer, H–3L × 4), 5.31 (dd, 0.4H, J 10.0, 10.0 Hz, H–3 from one isomer), 5.68 (dd, 0.6H, J 10.0, 10.0 Hz, H–3 from one isomer), 6.16 (d, 0.4H, J 8.5 Hz, NH from one isomer), 6.67 (d, 0.6H, J 7.5 Hz, NH from one isomer), 7.22–7.39 (m, 15H, Ar–H); 13C NMR (125 MHz, CDCl3): δ 14.15 (CH3), 22.73 (CH2), 25.07 (CH2), 25.09 (CH2), 25.19 (CH2), 25.27 (CH2), 25.35 (CH2), 25.74 (CH2), 29.19 (CH2), 29.27 (CH2), 29.35 (CH2), 29.42 (CH2), 29.49 (CH2), 29.57 (CH2), 29.62 (CH2), 29.68 (CH2), 29.73 (CH2), 29.75 (CH2), 31.96 (CH2), 34.17 (CH2), 34.48 (CH2), 34.52 (CH2), 34.67 (CH2), 37.81 (CH2), 38.16 (CH2), 39.19 (CH2), 39.27 (CH2), 40.86 (CH2), 41.33 (CH2), 45.01 (CH2N), 46.00 (CH2N), 47.16 (CH2N), 48.00 (CH2N), 55.03 (C–2 from one isomer), 56.43 (C–2 from one isomer), 61.95 (C–6 from one isomer), 62.39 (C–6 from one isomer), 67.24 (OCH2), 67.29 (OCH2), 68.36 (OCH2), 68.42 (OCH2), 69.60 (m, (PhCH2O)2P), 70.13 (C–3L), 70.69 (C–3L), 71.29 (C–3L), 71.78 (C–3L), 72.05 (C–3 from one isomer), 72.71 (C–3 from one isomer), 73.33 (PhCH2 from one isomer), 73.37 (PhCH2 from one isomer), 73.82 (C–5 from one isomer), 73.89 (C–5 from one isomer), 74.01 (d, J 5.0 Hz, C–4 from one isomer), 74.38 (d, J 5.0 Hz, C–4 from one isomer), 99.53 (C–1 from one isomer), 100.88 (C–1 from one isomer), 127.57 (CH-Ar), 127.91 (CH-Ar), 128.08 (CH-Ar), 128.29 (CH-Ar), 128.36 (CH-Ar), 128.52 (CH-Ar), 128.54 (CH-Ar), 128.56 (CH-Ar), 128.58 (CH-Ar), 128.60 (CH-Ar), 128.60 (CH-Ar), 135.55 (m, C-Ar), 137.96 (C-Ar), 138.04 (C-Ar), 169.76 (CO), 170.10 (CO), 170.18 (CO), 170.24 (CO), 170.43 (CO), 170.58 (CO), 170.67 (CO), 173.16 (CO), 173.18 (CO), 173.22 (CO), 173.26 (CO), 173.39 (CO), 173.42 (CO), 173.48 (CO), 174.04 (CO); HRESI–MS (m/z) Calcd for C143H249N2O21P [M+Na]+: 2384.8047, found: 2384.8032.
3.6 N-(2-Hydroxyethyl)-N-{2-[2-deoxy-4-O-phosphono-3-O-((R)-3-tetradecanoyloxytetradecanoyl)-2-((R)-3-tetradecanoyloxytetradecanamido)-β-D-glucopyranosyloxy]-ethyl}-(R)-3-tetradecanoyloxytetradecanamide (2) and N-[2-(4-hydroxybutyloxy)-ethyl]-N-{2-[2-deoxy-4-O-phosphono-3-O-((R)-3-tetradecanoyloxytetradecanoyl)-2-((R)-3-tetradecanoyloxytetradecanamido)-β-D-glucopyranosyloxy]-ethyl}-(R)-3-tetradecanoyloxytetradecanamide (4)
:1 → CHCl3/MeOH/H2O, 4:1:0.1) to afford 2 (23 mg, 44%) and 4 (12 mg, 21%). Both 2 and 4 were freeze dried from a dioxane-CHCl3 mixture (95:5) to give white fluffy solids.
For compound 2: Rf 0.34 (CHCl3/MeOH/H2O, 4:1:0.1); [α]22D −0.2 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3:CD3OD, 4:1): δ 0.75 (t, 18H, J 7.0 Hz, 6 × CH3), 1.08–1.27 (br m, 114, 57 × CH2 of lipid), 1.37–1.58 (br m, 12H, H–4L × 3, H–3L′ × 3), 2.10–2.27 (m, 8H, H–2L′ × 3, H–2L × 1), 2.29–2.71 (br m, 4H, H–2L × 2), 3.17–3.29 (br m, H–2, H–6a, H–6b), 3.47–3.68 (br m, 7H, H–5, ROCH2CH2NCH2CH2OH), 3.71–3.88 (m, 2H, ROCH2CH2NCH2CH2OH), 4.03–4.15 (br m, 1H, H–4), 4.30 (d, 0.6H, J 8.0 Hz, H–1 from one isomer), 4.45 (d, 0.4H, J 8.0 Hz, H–1 from one isomer), 4.89–5.16 (br m, 4H, H–3, H–3L × 3); HRESI–MS (m/z) Calcd for C94H178N2O18P [M − H]—1654.2843, found: 1654.2837.
For compound 4: Rf 0.40 (CHCl3/MeOH/H2O, 4:1:0.1); [α]22D −0.2 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3:CD3OD, 4:1): δ 0.75 (t, 18H, J 7.0 Hz, 6 × CH3), 1.20–1.45 (br m, 114, 57 × CH2 of lipid), 1.52–1.75 (br m, 12H, H–4L × 3, H–3L′ × 3), 1.81–2.03 (br, m, 4H, ROCH2CH2CH2CH2OH), 2.20–2.51 (br m, 8H, H–2L′ × 3, H–2L × 1), 2.29–2.71 (br m, 4H, H–2L × 2), 3.28–3.43 (br m, 3H, H–2, H–6a, H–6b), 3.45–4.06 (br m, 13H H–5, ROCH2CH2NCH2CH2OCH2CH2CH2CH2OH), 4.41–4.65 (br m, 2H, H–1, H–4), 5.02–5.33 (br m 4H, H–3, H–3L × 3); ESI–MS (m/z) Calcd for C98H186N2O19P [M − H]− 1726.3, found: 1726.3.
3.7 N-{2-[(R)-3-tetradecanoyloxytetradecanoyloxy]-ethyl}-N-{2-[2-deoxy-4-O-phosphono-3-O-((R)-3-tetradecanoyloxytetradecanoyl)-2-((R)-3-tetradecanoyloxytetradecanamido)-β-D-glucopyranosyloxy]-ethyl}-(R)-3-tetradecanoyloxytetradecanamide (3)
In a similar manner as described for the global deprotection of 12, a solution of 13 (12 mg, 0.005 mmol) in freshly distilled THF (30 mL) and palladium on charcoal (5%, 15 mg) was stirred at room temperature under hydrogen atmosphere for 24 h. The mixture was filtered, the filtrate concentrated, and the resulting residue purified by flash column chromatography (CHCl3/MeOH, 9:1 → CHCl3/MeOH/H2O, 4:1:0.1) to afford 3 (9 mg, 85%) as white fluffy solid after being freeze dried from a dioxane-CHCl3 mixture (95:5). Rf 0.54 (CHCl3/MeOH/H2O, 4:1:0.1); [α]22D −0.1 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3:CD3OD, 2:1): δ 0.82 (t, 24H, J 7.0 Hz, 8 × CH3), 1.13–1.34 (br m, 114, 57 × CH2 of lipid), 1.42–1.70 (br m, 16H, H–4L × 4, H–3L′ × 4), 2.17–2.43 (br m, 10H, H–2L′ × 4, H–2L × 1), 2.45–2.77 (br m, 6H, H–3L × 3), 3.22–3.31 (br m, 3H, H–2, H–6a, H–6b), 3.38–3.73 (br m, 7H, OCH2, CH2N × 2), 4.03–4.19 (br m, 2H, OCH2), 4.36–4.48 (br m, 2H, H–1, H–4), 4.89–5.08 (br m, 5H, H–3, H–3L × 4); HRESI–MS (m/z) Calcd for C122H230N2O21P [M − H]−: 2090.6746, found: 2090.6727.
3.8 Reagents for biological experiments
E. coli LPS 0111:B4 was obtained from InvivoGen, San Diego, CA. Each of synthetic lipid A mimics 2–4 was reconstituted in DMSO with brief sonication, aliquoted and stored at −80 °C. A fresh aliquot was used for each individual experiment. Solution concentrations were set such that the total addition of DMSO never exceed 0.5% to avoid toxic effects. THP-1 cells were obtained from American Type Culture Collection (ATCC). RPMI-1640 media, fetal bovine serum, and 1% antibiotic-antimycotic 100C were obtained from Gibco BRL. Phorbol 12-myristate 13-acetate (PMA) was purchased from Sigma, dissolved in DMSO, aliquoted and stored at −80 °C.3.9 Cell maintenance
THP-1 cells were maintained at 37 °C and 5% CO2 atmosphere in RPMI-1640 media supplemented with 10% heat-inactivated fetal bovine serum and 1% antibiotic-antimycotic 100X. Cell counting was performed using a Beckman Coulter ViCell X-R instrument, with viability being determined through the trypan blue cellular exclusion method.3.10 ICAM-1 induction and measurement
THP-1 cells were plated at 0.5 × 106 cells well−1 in 6-well tissue culture plates and incubated for 18 h. Cells were then incubated with different stimuli for a further 18 h. Cells were centrifuged at 1000 g for 5 min, washed with phosphate buffered saline (PBS), and then resuspended in 100 μL 0.1% Bovine Serum Albumin/PBS to proceed with staining and flow cytometry analysis. The expression of ICAM-1 was determined via immunostaining with phycoerythrin-conjugated mAb to ICAM-1 (CD54) from BD Biosciences, San Jose, CA. The antibody was added and the mixture incubated at 4 °C for 20 h in the dark. After incubation, cells were washed twice with PBS, resuspended in 500 μL PBS, and subjected to flow cytometry analysis on FACSCalibur with CELLQUEST PRO software (BD Biosciences), acquiring 15,000 events. The results were presented as the mean fluorescence intensity (MFI) on the FL2 channel.3.11 Cytokine induction and measurement:
THP-1 pre-monocytic cells were plated at 0.5 × 106 cells well−1 in 6-well tissue culture plates containing the RPMI media further supplemented with 5 ng mL−1 of phorbol 12-myristate 13-acetate (PMA). After 48 h, the media was removed and the now adherent monocytic THP-1 cells were washed with PBS. The wells were then refilled with serum-free RPMI media, incubated for 3 h, and then exposed to stimuli. After 24 h stimulation, culture supernatants were collected and stored frozen (−80 °C) until assayed for cytokine production.All cytokine ELISAs were performed in 96-well MaxiSorp plates. Ready-Set-Go! ELISA kits (eBioscience) were used for cytokine quantification of human TNF-α, IL-6, IL-1β according to the manufacturer's instructions. The absorbance was measured at 450 nm with wavelength correction set to 540 nm using a microplate reader (BMG Labtech). All cytokine values were measured in duplicate, and are presented as the mean ± SD of two separate experiments.
Acknowledgements
Compound 5 used for the synthesis of lipid A mimics (2–4) was a kind gift from Biomira Inc. This work was supported by Natural Sciences and Engineering Research Council of Canada (NSERC, Grant 312630) and Lakehead University.References
- C. R. Raetz and C. Whitfield, Annu. Rev. Biochem., 2002, 71, 635–700 CrossRef CAS.
- O. Westphal and O. Lüderitz, Z. Naturforsch., 1952, 7, 148–155 Search PubMed.
- K. Miyake, J. Endotoxin Res., 2006, 12, 195–204 CrossRef CAS.
- J. Gmeiner, O. Lüderitz and O. Westphal, Eur. J. Biochem., 1969, 7, 370–379 CrossRef CAS.
- S. Hase and E. Rietschel, Eur. J. Biochem., 1976, 63, 101–107 CrossRef CAS.
- S. Akira and M. Yamamoto, Adv. Exp. Med. Biol., 2010, 667, 59–68 Search PubMed.
- B. S. Part, D. H. Song, H. M. Kim, B. S. Choi and J. O. Lee, Nature, 2009, 458, 1191–1196 CrossRef.
- U. Ohto, K. Fukase, K. Miyake and Y. Satow, Science, 2007, 316, 1632–1634 CrossRef CAS.
- L. D. Hawkins, W. J. Christ and D. D. Rossignol, Curr. Top. Med. Chem., 2004, 4, 1147–1171 CrossRef CAS.
- Z.-H. Jiang and R. R. Koganty, Curr. Med. Chem., 2003, 10, 1423–1439 CrossRef CAS.
- R. J. Ulevitch, Nat. Rev. Immunol., 2004, 4, 512–520 CrossRef CAS.
- Y. Zhang, J. Gaekwad, M. A. Wolfert and G.-J. Boons, J. Am. Chem. Soc., 2007, 129, 5200–5216 CrossRef CAS.
- K. K. Maiti, M. Decastro, A. B. EI-Sayed, M. I. foote, M. A. Wolfert and G. J. Boons, Eur. J. Org. Chem., 2010, 80–91 CAS.
- J. Gaekwad, Y. Zhang, W. Zhang, J. Reeves, M. A. Wolfert and G. J. Boons, J. Biol. Chem., 2010, 285, 29375–29386 CrossRef CAS.
- S. Kusumoto, M. Hashimoto and K. Kawahara, Adv. Exp. Med. Biol., 2010, 667, 5–23 CrossRef.
- S. Kusumoto, K. Fukase, Y. Fukase, M. Kataoka, H. Yoshizaki, K. Sato, M. Oikawa and U. Suda, J. Endotoxin Res., 2003, 9, 361–366 CAS.
- D. A. Johnson, Curr. Top. Med. Chem., 2008, 8, 64–79 CrossRef CAS.
- A. G. Stöver, J. D. S. Correia, J. T. Evans, C. W. Cluff, M. W. Elliott, E. W. Jeffery, D. A. Johnson, M. J. Lacy, J. R. Baldridge, P. Probst, R. J. Ulevitch, D. H. Persing and R. M. Hershberg, J. Biol. Chem., 2004, 6, 4440–4449 Search PubMed.
- O. R. Martin, W. Zhou, X. Wu, S. Font-Deschamps, S. Moutel, K. Schindl, P. Jeandet, C. Zbaeren and J. A. Bauer, J. Med. Chem., 2006, 49, 6000–6014 CrossRef CAS.
- Z.-H. Jiang, W. A. Budzynski, L. N. Skeels, M. J. Krants and R. R. Koganty, Tetrahedron, 2002, 58, 8833–8842 CrossRef CAS.
- E. Lien, J. C. Chow, L. D. Hawkins, P. D. McGuinness, K. Miyake, T. Espevik, F. Susovsky and D. D. Golenbock, J. Biol. Chem., 2001, 276, 1873–1880 CrossRef CAS.
- J. Eustache, A. Grob and H. Retscher, Carbohydr. Res., 1994, 251, 251–267 CrossRef CAS.
- M. A. Bulusu, P. Waldstatten, J. Hildebrandt, E. Schutze and G. Schulz, J. Med. Chem., 1992, 35, 3463–3469 CrossRef CAS.
- K. Ikeda, T. Asahara and K. Achiwa, Chem. Pharm. Bull., 1993, 41, 1879–1881 CrossRef CAS.
- Z.-H. Jiang, W. A. Budzynski, L. N. Skeels, M. J. Krantz and R. R. Koganty, Tetrahedron, 2002, 58, 8833–8842 CrossRef CAS.
- Z.-H. Jiang, M. V. Bach, W. A. Budzynski, M. J. Krantz, R. R. Koganty and B. M. Longenecker, Bioorg. Med. Chem. Lett., 2002, 12, 2193–2196 CrossRef CAS.
- Z.-H. Jiang, W. A. Budzynski, D. Qiu, D. Yalamati and R. R. Koganty, Carbohydr. Res., 2007, 342, 784–796 CrossRef CAS.
- Z.-H. Jiang, R. Xu, C. Wilson and A. Brenk, Tetrahedron Lett., 2007, 48, 2915–2918 CrossRef CAS.
- M. Kiso, S. Tanaka, M. Fujita, Y. Fujishima, Y. Ogawa and A. Hasagawa, Carbohydr. Res., 1987, 162, 247–256 CrossRef CAS.
- Y. Matsuo, A. Iwashita, H. Oyama and E. Nakamura, Tetrahedron Lett., 2009, 50, 3411–3413 CrossRef CAS.
- G. A. Olah, Q. Wang, X. Li, G. Rasul and G. K. S. Prakash, Macromolecules, 1996, 29, 1857–1861 CrossRef CAS.
- I. S. Akhrem, D. V. Avetisvan, S. V. Vitt and P. V. Petrovskii, Mendeleev Commun., 2005, 15, 185–187 CrossRef.
- L. M. van Renterghem, E. J. Goethals and F. E. Du Prez, Macromolecules, 2006, 39, 528–534 CrossRef CAS.
- D. Stanssens, R. Hermanns and H. Wories, Prog. Org. Coat., 1993, 22, 379–391 CrossRef CAS.
- R. A. T. M. van Benthem, N. Neijerink, E. Geladé, C. G. de Koster, D. Muscat, P. E. Froehling, P. H. M. Hendriks, C. J. A. A. Vermsulen and T. J. G. Zwartkruis, Macromolecules, 2001, 34, 3559–3566 CrossRef CAS.
- Z. W. Jr. Wicks and N. C. J. Chiang, Coat. Technol., 1982, 54, 27–31 CAS.
- J. Lomax and G. J. Swift, Coat. Technol., 1982, 50, 49–55 Search PubMed.
- D. A. Johnson, C. G. Sowell, C. L. Johnson, M. T. Livesay, D. S. Keegan, M. J. Rhodes, J. T. Ulrich, J. R. Ward, J. L. Cantrell and V. G. Brookshire, Bioorg. Med. Chem. Lett., 1999, 9, 2273–2278 CrossRef CAS.
- M. D. Kalluri, P. Datla, A. Bellary, K. Basha, A. Sharma, A. Sharma, S. Singh, S. Upadhyay and V. Rajagopal, FEBS J., 2010, 277, 1639–1952 CrossRef CAS.
- S. H. Lee, H. K. Jun, H. R. Lee, C. P. Chung and B. K. Choi, Int. J. Antimicrob. Agents, 2010, 35, 138–145 CrossRef CAS.
- J. M. Luk, W. Lai, P. Tam and M. W. Koo, Life Sci., 2000, 67, 155–163 CrossRef CAS.
- K. A. Roebuck and A. Finnegan, J. Leukocyte Biol., 1999, 66, 876–888 CAS.
- A. Jahnke, A. van de Stolpe, E. Caldenhoven and J. P. Johnson, Eur. J. Biochem., 1995, 228, 439–446 CrossRef CAS.
- S. Takashiba, T. E. van Dyke, S. Amar, Y. Murayama, A. W. Soskolne and L. Shapira, Infect. Immun., 1999, 67, 5573–5578 CAS.
- E. K. Park, H. S. Jung, H. I. Yang, M. C. Yoo, C. Kim and K. S. Kim, Inflammation Res., 2007, 56, 45–50 CrossRef CAS.
- F. L. van de Veerdonk, M. G. Netea, C. A. Dinarello and L. A. Joosten, Trends Immunol., 2011, 32, 110–116 CrossRef CAS.
- C. A. Dinarello, Blood, 1996, 97, 2095–2147 Search PubMed.
- E. Latz, Curr. Opin. Immunol., 2010, 22, 28–33 CrossRef CAS.
- C. A. Salkowski, G. R. Detore and S. N. Vogel, Infect. Immun., 1997, 65, 3239–3247 CAS.
- K. Okemoto, K. Kawaski, K. Hanada, M. Miura and M. Nishijima, J. Immunol., 2006, 176, 1203–1208 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra01149b |
| This journal is © The Royal Society of Chemistry 2012 |