Ligand selection from the analysis of protein conformational substates: new leads targeting the N-terminal domain of Hsp90
Alessandro
Genoni
ab,
Marzia
Pennati
c,
Giulia
Morra
a,
Nadia
Zaffaroni
c and
Giorgio
Colombo
*a
aIstituto di Chimica del Riconoscimento Molecolare, CNR, Via Mario Bianco 9, 20131, Milano, Italy. E-mail: giorgio.colombo@icrm.cnr.it; Fax: +39-02-28901239; Tel: +39-02-28500031
bEquipe de Chimie et Biochimie Théoriques, UMR 7565 SRSMC—CNRS, Université de Lorraine, Boulevard des Aiguillettes BP 70239, 54506, Vandoeuvre-lès-Nancy Cedex, France
cDepartment of Experimental Oncology and Molecular Medicine, Fondazione IRCCS Istituto Nazionale dei Tumori, Via Amadeo 42, 20133, Milano, Italy
First published on 28th March 2012
Abstract
The dynamic properties of proteins underlie every aspect of their functions in the cell. The atomistic description of protein motions and their inclusion in ligand selection processes may provide new opportunities for hit identification and drug discovery. Herein, we present a novel rational strategy that allowed us to computationally select new N-terminal targeted inhibitors of the molecular chaperone Hsp90 starting from the atomistic analysis of the conformational dynamics of the complex between the protein and its natural ligand ATP. First of all, we have identified the relevant representatives of distinct conformational substates of the Hsp90–ATP complex through the application of a novel structural clustering strategy and, for each of them, we have afterwards characterized the nucleotide–protein interactions to build a pharmacophore model recapitulating the binding hotspots conserved in different ensembles of protein conformations. The resulting pharmacophore has been finally used to screen a database of small molecules and allowed us to identify novel drug-like molecules with interesting activities against Hsp90 functions in experimental models of cancer cells. The results and the experimental validation of the selected molecules provide support for the feasibility of including protein flexibility in drug selection strategies through the characterization of relevant substates.
Introduction
Proteins are inherently flexible systems in constant motion among different conformational substates and their dynamic properties are frequently associated with molecular recognition, function and catalysis. However, internal motions and the concept of conformational substates have often been ignored in classical structure-based drug discovery despite recent successes in the design of small-molecule inhibitors.1–7The role of functional dynamics has often been conceptualized in terms of a model in which the protein pre-exists in an ensemble of energetically similar conformations. In this context, conformational selection is defined as a process in which a ligand selectively binds to one (or a subset) of these conformers, stabilizing it and increasing its relative population with respect to the total protein population, eventually resulting in the final observed complex. The concepts of ligand-induced shifts in protein conformational dynamics can be usefully exploited in the context of drug design and discovery. In fact, in some cases ligand binding induces specific conformational states that might not be anticipated from the simple analysis of the protein apo structure and may even result in major structural transitions.
One notable instance of ligand-regulated protein functional dynamics is represented by Heat shock protein 90 (Hsp90). Hsp90 is an ATPase-directed molecular chaperone, operating in a complex cycle driven by ATP binding, hydrolysis, and ATP/ADP exchange at the N-terminal nucleotide-binding site.8 The protein is a homodimer in which each protomer is characterized by a modular architecture with three domains: an N-terminal regulatory domain (NTD), responsible for ATP binding, a middle domain (M-domain), which completes the ATPase site necessary for ATP hydrolysis and also binds client proteins, and a C-terminal domain (CTD)9 required for dimerization. Crystal structures, solution and biochemical studies suggest a role for the nucleotide in selecting and stabilizing different conformations of Hsp90, related to specific functions in the chaperone cycle.10,11 Overall, the mechanism of conformational coupling to the ATPase cycle appears to involve a “tense” and closed conformational state of Hsp90 upon ATP binding, whereas subsequent hydrolysis to ADP leads to a more “relaxed” and structurally flexible state of Hsp90. Finally, in the nucleotide-free form, the dimer moves to an “open” state.9 Given its importance in cancer development and maintenance, Hsp90 inhibition has been intensely pursued in recent years. Thirteen inhibitors, all targeting the N-terminal ATP-binding pocket, have entered clinical trials.12
In this paper, we present a novel rational strategy for the computational discovery of N-terminal targeted inhibitors of Hsp90, which is based on the explicit inclusion of the flexibility and conformational dynamics of the active complex between the chaperone and its natural ligand ATP.
Our approach starts from the identification of the relevant representatives of distinct conformational substates visited during a long timescale molecular dynamics (MD) simulation of the complex between full length Hsp90 and ATP. The analysis of the interactions between the nucleotide and the binding site and their fluctuations within different substates is used to build a pharmacophore model in which the relative importance of each pharmacophoric feature is weighted according to the conservation of the interaction it mimics in the different substates. The resulting pharmacophore takes into account in a simple and compact manner the effects of protein mobility on ligand binding and the reciprocal effect of the nucleotide on the internal dynamics of the protein. Afterwards, the pharmacophore is used to screen a database of small molecules with the aim of identifying novel antagonists of the Hsp90 chaperone function with possible anticancer activity. It is important to underline that the pharmacophore model proposed here relies mainly on mimicking hydrogen bond interactions within the active site. Since this approximation might lead to the discovery of ligands that may have non-optimal drug-like properties, we have alleviated this problem by filtering the first selection of compounds through the application of the set of rules introduced by Lipinski.13
The results and the experimental validation of the activities of the selected molecules provide support to the feasibility of including flexibility in drug selection strategies and give a vivid illustration of how the knowledge of the internal dynamics of a system can be exploited to expand the molecular diversity space of possible ligands.
Materials and methods
Molecular dynamics studies for the selection of relevant ATP-induced substates
The MD simulation trajectories used in this work were obtained as described in a previously published paper.14 The details and the full description of the MD set up and runs can thus be found in that published paper, which deals with the characterization of the ligand-modulation of Hsp90 internal dynamics.14Briefly, the crystal structure9 (pdb entry 2CG9) containing the yeast Hsp90 dimer bound to ATP was employed as a starting point for the simulations. The system was solvated in a tetrahedral solvation box containing around 57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 particles. The simulations and the analysis of the trajectories were performed using the GROMACS software package15 using the GROMOS96 force field16 and the SPC water model.17 In order to remove possible bad contacts in the initial structure, the ATP-bound Hsp90 dimer system was initially optimized by means of a 2000-step steepest descent energy minimization and afterwards relaxed by a further 2000-step conjugate gradient energy optimization. The system was then equilibrated by a 50 ps MD run with position restraints on the protein and ligand to allow the relaxation of the solvent molecules, equilibration that was followed by another 50 ps MD run without position restraints on the solute. It is worthwhile to note that all the trajectory equilibrations were checked by monitoring quantities such as the RMSD with respect to the initial structure, the internal protein energy and the fluctuations calculated at different time-intervals. Furthermore, in all the performed MD simulations we took into account the long-range Coulomb interactions exploiting the particle mesh Ewald summation method,18,19 we used the LINCS algorithm20 to constrain all the bond lengths and the SETTLE program to deal with the water molecules.21 A dielectric permittivity, ε = 1, and a time step of 2 fs were also used. All atoms were given an initial velocity obtained from a Maxwellian distribution at the desired initial temperature of 300 K. The density of the system was adjusted by performing the first equilibration runs under NPT conditions by weak coupling to a bath of constant pressure (P0 = 1 bar, coupling time τP = 0.5 ps).22 In all the simulations the temperature was maintained close to the intended values by weak coupling to an external temperature bath22 with a coupling constant of 0.1 ps. The proteins and the rest of the system were coupled separately to the temperature bath.
000 particles. The simulations and the analysis of the trajectories were performed using the GROMACS software package15 using the GROMOS96 force field16 and the SPC water model.17 In order to remove possible bad contacts in the initial structure, the ATP-bound Hsp90 dimer system was initially optimized by means of a 2000-step steepest descent energy minimization and afterwards relaxed by a further 2000-step conjugate gradient energy optimization. The system was then equilibrated by a 50 ps MD run with position restraints on the protein and ligand to allow the relaxation of the solvent molecules, equilibration that was followed by another 50 ps MD run without position restraints on the solute. It is worthwhile to note that all the trajectory equilibrations were checked by monitoring quantities such as the RMSD with respect to the initial structure, the internal protein energy and the fluctuations calculated at different time-intervals. Furthermore, in all the performed MD simulations we took into account the long-range Coulomb interactions exploiting the particle mesh Ewald summation method,18,19 we used the LINCS algorithm20 to constrain all the bond lengths and the SETTLE program to deal with the water molecules.21 A dielectric permittivity, ε = 1, and a time step of 2 fs were also used. All atoms were given an initial velocity obtained from a Maxwellian distribution at the desired initial temperature of 300 K. The density of the system was adjusted by performing the first equilibration runs under NPT conditions by weak coupling to a bath of constant pressure (P0 = 1 bar, coupling time τP = 0.5 ps).22 In all the simulations the temperature was maintained close to the intended values by weak coupling to an external temperature bath22 with a coupling constant of 0.1 ps. The proteins and the rest of the system were coupled separately to the temperature bath.
The production MD simulation was performed for 170 ns, but the fist 5 ns were not considered in order to minimize convergence artefacts. The resulting trajectory was afterwards analyzed and partitioned according to the clustering procedure that has been recently introduced by Micheletti and coworkers23 and that will be briefly described in the following subsection.
Clustering of the trajectory for the definition of functional substates
The clustering procedure adopted in our work to identify the relevant representatives of distinct conformational substates from an MD simulation is based on the K-medoids clustering scheme.24 This technique considers as the input only the pairwise RMSD distances between all the pairs of recorded structures and provides a pre-assigned number K of non-empty and homogeneous clusters, each of them characterized by a representative conformation. The method identifies the clusters iteratively minimizing the “dissimilarity score” obtained by summing the RMSD of each structure from its cluster representative and, in particular, consists in the following three steps:1) random assignment of the members to the K clusters;
2) identification of the representative for each cluster as the element with the smallest total distance from the other cluster members;
3) redefinition of the clusters assigning each structure to the closest representative.
Steps 2 and 3 are iterated until the “dissimilarity score” converges to a minimum, but it is worthwhile to note that, in order to avoid trapping in local minima, the whole procedure is consistently repeated considering several starting random clusters.
It is obvious that the standard K-medoids technique24 relies only on geometrical considerations (namely, the RMSD distance of any pairs of structures), but it does not take into account any time-information, such as the time-succession of the recorded conformations in the examined trajectory. Time-information is fundamental to partition the trajectory in exam in a series of progressively visited substates and, therefore, in order to overcome this drawback Micheletti and coworkers have recently devised a modified K-medoids scheme to ensure that all the structures assigned to a cluster span a continuous time-interval.23 Of course, the time-continuity constraint simplifies the definition of the cluster members because they are simply specified by K − 1 time-partitions of the trajectory. In particular, the new modified technique starts with an equally spaced K − 1 subdivision of the simulation time-length and the representative of each cluster is afterwards determined as in step 2 of the standard technique. After calculating the associated “dissimilarity score”, a new clustering is proposed by randomly reassigning one or more of the K − 1 subdivisions with the only constraint that there are not identical subdivisions that would result in empty clusters. At this point the new cluster representatives can be identified and a new “dissimilarity score” can be computed. Following a Monte Carlo philosophy, the proposed clustering is accepted if it is associated with a lower “dissimilarity score”, otherwise the previous one is retained and another new partition is randomly proposed. The procedure is usually repeated until convergence of the “dissimilarity score”.
Dynamic pharmacophore design
In order to design a dynamic pharmacophore, first of all, we have performed the analysis of the conserved hydrogen bonds in all the relevant states of the Hsp90–ATP complex, selected according to the clustering procedure described above. This analysis has been performed exploiting the g_hbond utility of the GROMACS simulation package.15 We assumed the following geometric criteria to identify the hydrogen bonds: 1) the distance between the hydrogen atom and the acceptor atom is lower than 3.5 Å; 2) the angle formed by the donor atom, the hydrogen atom and the acceptor atom is greater than 120°; 3) the angle formed by the hydrogen atom, the acceptor atom and the acceptor-antecedent atom (namely, an atom one covalent bond away from the acceptor) is greater than 90°. As we will explain in the “Results” section, the hydrogen bond analysis allowed us to construct the pharmacophore ATP-PHARM-1 model (see Fig. 2) using the module to create pharmacophore queries in the DISCOVERY STUDIO 2.5 package.25 In particular we created a ten-point model by assembling eight hydrogen bond acceptors mapped over the OB, O5, O8, OA, O9, OC, O3 and O6 atoms of the ATP molecule (see Fig. 1) and two hydrogen bond donors mapped over the ATP groups OC–H5 and N4–H1 (see Fig. 1). Furthermore, we assigned a weight to all these pharmacophore features according to the overall occurrence of the corresponding hydrogen bond descriptors in the selected Hsp90–ATP complexes (see Tables 1 and 2). | ||
| Fig. 1 ATP molecule with labels for atoms considered in the hydrogen-bond analyses for the construction of the pharmacophore model. | ||
 | ||
| Fig. 2 The ten-feature pharmacophore model ATP-PHARM-1 plotted on the ATP molecule. Green spheres and arrows represent hydrogen bond acceptors, while purple spheres and arrows represent hydrogen bond donors. This is the pharmacophore used for screening the small molecule NCI_3D database. | ||
| HB Acceptor | Occurrence | Relative weight | Presence in the pharmacophore model |
|---|---|---|---|
| OB | 28 | 2.00 | Yes |
| O5 | 19 | 1.35 | Yes |
| O8 | 19 | 1.35 | Yes |
| OA | 19 | 1.35 | Yes |
| O9 | 11 | 0.79 | Yes |
| OC | 10 | 0.71 | Yes |
| O3 | 8 | 0.57 | Yes |
| O6 | 8 | 0.57 | Yes |
| O0 | 5 | 0.36 | No |
| O2 | 3 | 0.21 | No |
| O7 | 3 | 0.21 | No |
| O1 | 2 | 0.14 | No |
| O4 | 0 | 0.00 | No |
| N2 | 0 | 0.00 | No |
| N3 | 0 | 0.00 | No |
| N5 | 0 | 0.00 | No |
| HB Acceptor | Occurrence | Relative weight | Presence in the pharmacophore model |
|---|---|---|---|
| OC–H5 | 11 | 0.79 | Yes |
| N4–H1 | 6 | 0.43 | Yes |
| O3–H4 | 3 | 0.21 | No |
| O2–H3 | 2 | 0.14 | No |
Small-molecule database screening and hit identification
The obtained pharmacophore model has been considered as a query in a search of the NCI_3D database. This search has been performed by means of the DISCOVERY STUDIO 2.5 package setting the “SEARCH” method equal to “BEST” and the “LIMIT” option equal to “BEST N”. This returned 294 ligands that have been further processed accepting only those compounds fulfilling three of the four Lipinski rules.13 In fact, for the sake of completeness, it is important to be reminded that the Lipinski rule of five13 states that a molecule, to potentially be an orally active drug, must fulfil three of the following four criteria: a) no more than five hydrogen bond donors; b) no more than 10 hydrogen bond acceptors; c) a molecular mass lower than 500 daltons; d) an octanol–water partition coefficient logP not greater than 5. Actually, it is worthwhile to observe that we have used modified Lipinski rules since the desired molecules have been required to have a mass not greater than 550 daltons instead of 500 daltons. The screening using the modified Lipinski rules has been performed using the InstantJChem software26 and returned 25 compounds.Small molecules
Compounds for this study were kindly provided by the National Cancer Institute Repository (Bethesda, MD, USA). The 17-(allylamino)-17-demethoxygeldanamycin (17-AAG) was purchased from Sigma Aldrich (St. Louis, MO, USA). To obtain a 100 mM stock solution of the molecules, a variable amount of a solution of 1% dimethyl sulfoxide (DMSO) made in sterile water was added to each powdered stock and the compound was completely dissolved, stored at −20 °C, and diluted in complete culture medium, at the appropriate working solution, immediately before use.Evaluation of the antiproliferative potential of small molecules
4 × 103 cells were plated in 96-well flat-bottomed microtiter plates for 24 h and then treated with increasing concentrations of different compounds (0.01–50 μM) for 72 h. Control cells received vehicle alone (1% DMSO). All studies were performed in eight replicates and repeated at least three times independently. At the end of drug exposure, the antiproliferative potential was determined with the sulforhodamine B (SRB) protein staining method as previously reported.27 Briefly, cells were fixed with cold 50% trichloroacetic acid solution (Sigma Aldrich) and stained with 0.4% SRB (Sigma Aldrich) dissolved in 1% acetic acid (Sigma Aldrich) for 30 min. The plates were then washed four times with 1% acetic acid to remove unbound stain and air-dried. The dye was then dissolved by 10 mM Tris base (pH 10.5). The optical density was read at 550 nm on a microplate reader and the results were expressed as a percentage, relative to DMSO-treated cells. Dose-response curves were created and IC50 and IC80 values (i.e., concentrations able to inhibit cell growth by 50% and 80%, respectively) were determined graphically from the curve for each compound.Analysis of Hsp90 chaperone function
To monitor changes in Hsp90 client proteins, at the end of each treatment, cells were scraped, pelleted, washed twice with PBS, and then solubilized in lysis buffer (0.01% NP40, 10 mM Tris [pH 7.5], 50 mM KCl, 5 mM MgCl2, 2 mM DTT, 20% glycerol plus protease inhibitors). Samples containing 40 μg of protein were resolved on precast 12% NuPAGE™ bis-tris gels (Invitrogen Life Technology, Camarillo, CA, USA), and were transferred onto Hybond ECL nitrocellulose membranes (GE Healthcare, Chalfont St. Giles, UK) using the NuPAGE transfer buffer (Invitrogen Life Technology). Nitrocellulose membranes were blocked in PBS-Tween 20 with 5% skim milk, first incubated with the primary antibodies specific for survivin (ab469) (Abcam Inc., Cambridge, UK), Hsp90 (sc7947), Hsp70 (sc24), CDK4 (sc136241), CDK6 (sc271364), (Santa Cruz Biotechnology, Santa Cruz, CA), and AKT (9272) (Cell Signaling Technology, Danvers, MA) and then with the secondary peroxidase-linked whole antibodies (GE Healthcare). Bound antibodies were detected using the SuperSignals West PICO chemiluminescent substrate (Pierce Biotechnology Inc., Rockford, IL, USA). β-Actin monoclonal antibodies (Abcam Inc.) were used to confirm equal protein loading on the gel.Cell cycle phase distribution and apoptosis analysis
After treatment with small molecules, both adherent and floating cells were fixed in 70% ethanol. Fixed cells were stained for 30 min with propidium iodide (50 μg ml−1) and RNase (50 mg ml−1) in PBS buffer. Samples were analyzed with a FACScan flow cytometer (Becton Dickinson, Sunnyvale, CA, USA) with an excitation wavelength of 488 nm and an emission wavelength of 585 nm. At least 30000 events were read and histograms were analyzed using the CellQuest software according to the Modfit model (Becton Dickinson).
For apoptosis analysis, an aliquot of propidium-stained cells was observed for nuclear morphology of apoptosis (chromatin condensation and DNA fragmentation) by fluorescence microscopy. The percentage of cells with an apoptotic nuclear morphology was determined by scoring at least 500 cells in each sample.
Statistical analysis
Two-tailed Student's t test was used for statistical analysis. P values <0.05 were considered statistically significant.Analysis of the molecular interactions established by the most promising hits with Hsp90
In order to gain an insight into the observed Hsp90 inhibitory/cellular activity of some of the most promising hits that resulted from the performed experimental tests, we have analyzed the molecular interactions that they establish with Hsp90. To accomplish this task we have docked each considered compound into the ATP binding site of all the Hsp90 conformations previously taken into account for the pharmacophore construction (namely, fourteen structures). In fact, in the light of what already stressed above, considering only one conformer would be rather limiting and we believe that examining several conformations would help us to unravel the way in which an inhibitor adapts to/influences the receptor dynamics. For the sake of completeness, it is worth mentioning that all the docking calculations were performed using the package AutoDock 4.2.28 In particular, grid maps were generated by the program AutoGrid with a 0.375 Å spacing, with a grid center corresponding to the position of the ATP-C7 atom (see Fig. 1) in the Hsp90-ATP complex of the dynamic substate under examination (namely, the grid was centered over the ATP binding site) and with 55 points in each directions (namely, 166375 points in total). Polar hydrogen atoms and Gasteiger charges were properly taken into account for the receptor and, furthermore, the Lennard–Jones parameters 12–10 and 12–6 were used to model hydrogen bonds and van der Waals interactions, respectively. Considering the real docking calculations, random orientations and torsions were used for each ligand and the number of active torsions was set equal to the maximum number of rotatable bonds. The Lamarckian genetic algorithm was used with the default docking parameters. Moreover, for each run, the population size, the maximum number of energy evaluations and the maximum number of generations were set equal to 150 million, 50 million and 27000, respectively. A total of 250 runs were performed. At the end of the docking runs, the conformations of the ligand were listed in an increasing energy order and the ligand conformation with the lowest binding energy was afterwards considered as reference. All the structures with an RMSD value lower than 2.0 Å from the reference structure were assigned to the first cluster and it is important to note that after a conformation is assigned to a cluster it was not used again for other energetically less-favorable clusters. The procedure was repeated for all the remaining unclassified structures until all of them were put in a cluster.
After each docking calculation, we have extracted the complex corresponding to the lowest binding energy conformation and we have analyzed the associated intermolecular interactions. Repeating this protocol for all the considered receptor conformations we were able to perform a statistical analysis of the Hsp90–inhibitor contacts and, finally, we detected those interactions that best conserve and stabilize the complex by also considering the protein fluctuations (see the “Results” section for more details).
Results
Identification of relevant conformational substates from the dynamics of the Hsp90–ATP complex
The conformational dynamics of Hsp90 in complex with ATP, representing the activated state of the chaperone, was characterized by a long time scale (170 ns) all-atom Molecular Dynamics simulation as already described above and by Morra et al.14 The trajectory was afterwards analyzed with a novel structural clustering scheme developed by Micheletti and coworkers23 to identify the relevant visited conformational substates. The details of the clustering procedure and of the identification of the substates are reported in the “Materials and Methods” section. The aim of this step was to select a minimal number of key configurations that capture most of the structural fluctuations encountered in the multi-nanosecond trajectory, recapitulating the flexibility of the complex and providing a compact representation of how internal dynamics reverberates on the types and persistence of intermolecular interactions between Hsp90 and ATP in the N-terminal domain.It is important to underline that the structural clustering strategy used herein accounts for the time-order of the structures, which is essential for partitioning the trajectory in a succession of progressively-visited and coherent substates. The time-continuity of the members of the different clusters is instrumental to simplifying the definition of the different homogeneous structural sets of the Hsp90–ATP complex that have been afterwards used as templates for pharmacophore design.
The clustering was performed by varying K from 2 to 15 and enforcing the time continuity of the clustered structures, according to the scheme described in the “Materials and Methods” section. The algorithm returned a subdivision of the trajectory into a fixed number K of non-overlapping intervals (clusters) with maximal internal structural homogeneity. An optimal subdivision was obtained using seven (namely, K = 7) non-overlapping intervals and it was consequently used to identify the most prominent conformational substates explored by the trajectory.
During the free dynamic evolution, the chaperone–nucleotide complex populates seven distinct conformational substates with residence times from 21.2 to 30.2 ns. The average RMSD between pairs of structures within each identified substate was 2 Å. In contrast, the ensemble of different conformers is structurally heterogeneous (with inter-substate differences of up to 5 Å), reflecting the well-known flexibility that characterizes the members of the Hsp90 family.
Dynamic pharmacophore design
As briefly mentioned in the introduction, in order to design a dynamic pharmacophore model for the discovery of new possible Hsp90 inhibitors, we have identified the most recurring fundamental interactions between Hsp90 and the ATP molecule, assigning them a weight according to the frequency with which they occur.In order to accomplish this task, first of all, we have assumed that the intermolecular interactions needed to stabilize the Hsp90–ATP complexes are mainly hydrogen bond-like interactions, which are directional interactions typically also found in drug-like molecules, and, therefore, we have selected all the possible hydrogen bond acceptors and donors (also indicated as potential hydrogen bond descriptors) in ATP (see Tables 1–2 and Fig. 1). We have afterwards considered the seven key configurations that we have previously selected by means of the clustering procedure described above, each corresponding to two protein–nucleotide complexes. For all of these complexes we have determined which are the potential hydrogen bond descriptors able to establish significant interactions in the ATP binding site, namely, those potential ATP hydrogen bond acceptors and donors that form effective hydrogen bonds with Hsp90 (see the “Materials and Methods” section for the details). We have thus determined the number of times each ATP hydrogen bond descriptor plays a fundamental role in substrate binding (see Tables 1 and 2) and we have afterwards divided these numbers by the number of analyzed Hsp90–ATP complexes (fourteen) in order to obtain the relative weights of all the hydrogen bond acceptors and donors in the pharmacophore definition (see Tables 1 and 2). Nevertheless, the ATP hydrogen bond descriptors with a weight lower than 0.4 have been discarded and this has led to the construction of the pharmacophore model labeled as ATP-PHARM-1 and represented in Fig. 2.
It is worth noting that a pharmacophore model such as the one proposed here, namely a model mainly based on reproducing hydrogen bond interactions, may select ligands with charged functionalities, a feature that potentially provides leads that are poor drug candidates. On the other hand, this rationally designed model may provide useful information for the selection of potentially active molecules with new structural and stereoelectronic properties. Moreover, given the directionality of the hydrogen bond interactions used in the pharmacophore, selected hits may display an optimal 3D organization of the functional groups needed to productively bind the ATP site of Hsp90. To evaluate the potentials and pitfalls of this model, an analysis of how known inhibitors fit to or diverge from our pharmacophore constraints was carried out. Therefore, we considered the crystal structures of the Hsp90 N-terminal domain with some known inhibitors: geldanamycin,29 which is included in the NCI database, radicicol,30 purine scaffold-based inhibitors31 (PU-H71, PU-H64 and PU24FCl) and a benzamide tetrahydro-4H-carbzol-4-one analog that represents a Serenex-type compound.32 From this analysis it results that whereas radicicol, PU-H71, PU-H64 and the Serenex-like inhibitor mainly establish only one important direct hydrogen bond with the residue Asp 79 (from now on we will use the residue numbering of the 2CG9 pdb file), the network of direct hydrogen bonds is much more extended for geldanamycin and PU24FCl. In particular, geldanamycin forms a hydrogen bond with Asp 79 through its carbamate amino group, which mimics the exocyclic –NH2 group of the ATP/ADP adenine, two crucial hydrogen bonds with Lys 44 by means of the oxygen atom of its hydroxyl group and the oxygen atom of the “C29” methoxy group,29 two hydrogen bonds with Gly 123 and Phe 124 through the ansa-ring carbonyl oxygen, which is the analog of the ATP/ADP α-phosphate group, and, finally, a hydrogen bond with Lys 98 by means of the benzoquinone oxygen atom, which mimics the ATP/ADP β-phosphate group. For the sake of completeness, we observe that PU24FCl forms two hydrogen bonds with Tyr 125 in addition to the fundamental hydrogen bond with Asp 79. From these last two examples, and especially from the geldanamycin case, we can conclude that our hydrogen bond based pharmacophore model partially reproduces the binding determinants of these two major inhibitors of Hsp90.
At this point, it is worth noting that one of the goals of our work was to select molecular entities that are different from known inhibitors (see the “Discussion” section). Moreover, the pharmacophore was designed using the concept of dynamics-based weighting the Hsp90–ATP interactions. As a consequence, it is not surprising that, compared to already known inhibitors, the model shows differences in the fine details of the molecular recognition determinants that it mimics.
Small-molecule database screening and hit identification
The new dynamic pharmacophore model was used to perform a screening search of the small molecule database NCI_3D that contains approximately 160000 compounds. Filtering with the pharmacophore returned 294 hits that were subject to a further screening constraint consisting of the fulfilment of three of the four Lipinski rules13 (see the “Materials and Methods” section for the details). This resulted in 25 molecules (see Fig. 3–5) that approximately correspond to 0.016% of the reference database. Despite the presence of charged functionalities, the overall properties of most of the selected compounds are consistent with drug-like features, since the second screening imposed the fulfilment of the Lipinski rules.13 To better investigate this aspect we have also explicitly calculated the logP values of the 25 hits (see Table 3). All the identified compounds fulfil the rule for the octanol–water partition coefficient (logP is lower than or equal to 5) even if most of them show negative values. However, a stricter extension of the Lipinski rules, which was proposed by Ghose and coworkers to better evaluate the druglikeness of molecules,33 defines the logP values as acceptable if they are between −0.4 and 5.6. As shown in Table 3, eight of our 25 hits fulfil the Ghose rule for the partition coefficient. More importantly, three members of this subset correspond to compounds that were shown to be active in experimental tests (see the next subsection).
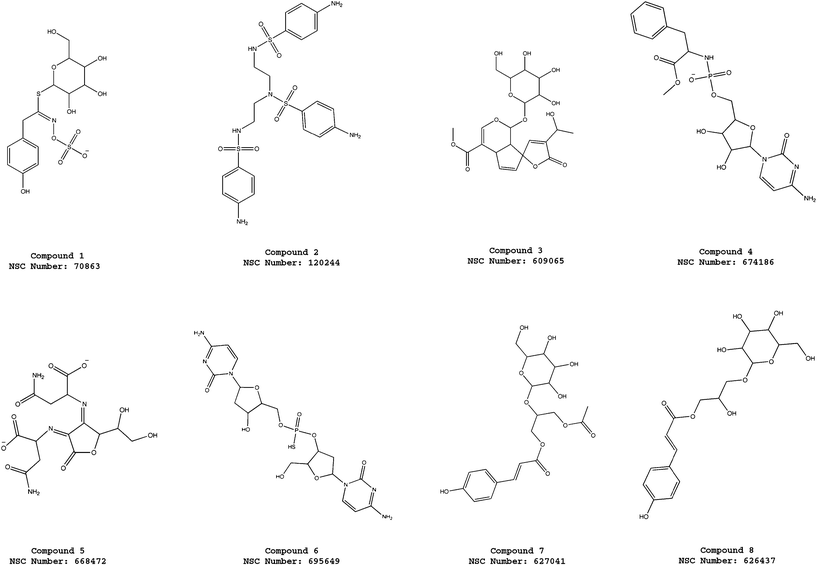 | ||
| Fig. 3 Molecular structures of the compounds 1–8 selected from the virtual screening of the NCI_3D database using the pharmacophore model ATP-PHARM-1. | ||
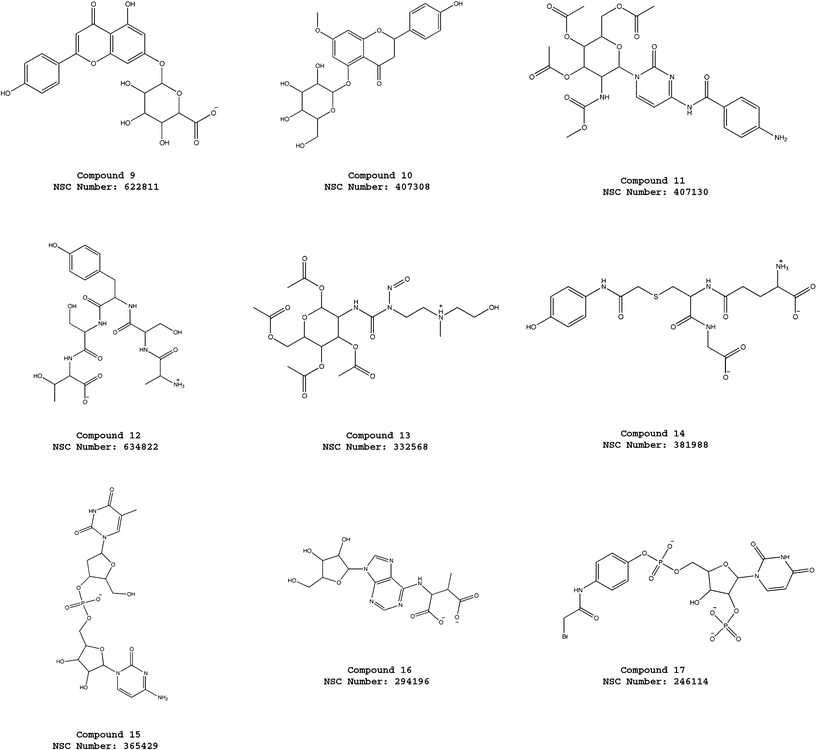 | ||
| Fig. 4 Molecular structures of the compounds 9–17 selected from the virtual screening of the NCI_3D database using the pharmacophore model ATP-PHARM-1. | ||
 | ||
| Fig. 5 Molecular structures of the compounds 18–25 selected from the virtual screening of the NCI_3D database using the pharmacophore model ATP-PHARM-1. | ||
| Compound | logP | Lipinski | Ghose | Activity |
|---|---|---|---|---|
| 1 | −2.58 | Yes | No | TBD |
| 2 | −0.20 | Yes | Yes | Yes |
| 3 | −1.64 | Yes | No | TBD |
| 4 | −1.57 | Yes | No | Yes |
| 5 | −3.00 | Yes | No | No |
| 6 | −2.94 | Yes | No | TBD |
| 7 | −0.44 | Yes | No | TBD |
| 8 | −0.88 | Yes | No | TBD |
| 9 | +0.76 | Yes | Yes | TBD |
| 10 | +0.06 | Yes | Yes | No |
| 11 | −0.82 | Yes | No | No |
| 12 | −6.36 | Yes | No | TBD |
| 13 | −1.63 | Yes | No | TBD |
| 14 | −4.22 | Yes | No | TBD |
| 15 | −3.55 | Yes | No | TBD |
| 16 | −3.30 | Yes | No | TBD |
| 17 | −0.67 | Yes | No | TBD |
| 18 | −3.75 | Yes | No | TBD |
| 19 | −0.10 | Yes | Yes | Yes |
| 20 | +2.16 | Yes | Yes | Yes |
| 21 | +0.16 | Yes | Yes | TBD |
| 22 | −1.80 | Yes | No | TBD |
| 23 | −6.89 | Yes | No | No |
| 24 | +0.98 | Yes | Yes | TBD |
| 25 | +0.90 | Yes | Yes | TBD |
For the sake of completeness, it is important to note that the screening with our pharmacophore model was not able to select geldanamycin, which is part of the NCI database. This is due to the fact that our pharmacophore captures only some of the binding determinants that characterize the macrocyclic inhibitor. Nevertheless, we want to stress again that one of the aims of our work was to develop and test a new pharmacophore design strategy allowing the discovery of new active molecules against Hsp90, possibly different from already identified active compounds.
Experimental tests on the newly discovered Hsp90 inhibitors
Eight of the selected compounds (2, 4, 5, 10, 11, 19, 20, 23) resulting from the virtual screening were obtained from the National Cancer Institute (NCI) and tested in a panel of human breast carcinoma cell lines. Cells were exposed for 72 h to increasing concentrations (from 0.5 to 50 μM) of the small molecules, and the effect on cell proliferation was determined by SRB assay. Assessment of the IC50 values showed that only compounds 2, 4, 19 and 20 inhibited cell growth in a concentration-dependent manner. However, while compounds 2 and 19 exhibited a comparable potency in all tumor cell lines, compounds 4 and 20 induced a variable growth inhibitory effect in the different cell models (see Table 4). At tested drug concentrations, IC50 was not reached for the other compounds (5, 11, 10 and 23) (see Table 4). For comparative purposes, the antiproliferative effect of 17-AAG was also evaluated and IC50 values ranging from 0.08 ± 0.01 μM to 3.48 ± 0.22 μM were observed for the different cell lines (see Table 4).| Cell Line | IC50 values (μM) for the different compounds | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 2 | 4 | 5 | 10 | 11 | 19 | 20 | 23 | 17-AAG | |
| BT-474 | 27.3 ± 4.8 | 27.1 ± 3.6 | >50 | >50 | >50 | 24.1 ± 4.8 | 21.5 ± 3.4 | >50 | 0.23 ± 0.02 |
| HCC1937 | 37.3 ± 4.4 | 27.8 ± 4.2 | >50 | >50 | >50 | 22.2 ± 3.4 | 37.2 ± 5.6 | >50 | 0.08 ± 0.01 |
| MCF-7 | 21.3 ± 2.8 | 4.2 ± 1.1 | >50 | >50 | >50 | 15.1 ± 2.1 | 43.1 ± 2.8 | >50 | 3.48 ± 0.22 |
| MDA-MB-231 | 25.1 ± 3.2 | 6.3 ± 1.2 | >50 | >50 | >50 | 19.3 ± 2.8 | 16.8 ± 2.9 | >50 | 1.12 ± 0.04 |
| T-47D | 25.3 ± 1.7 | 2.6 ± 0.8 | >50 | >50 | >50 | 16.4 ± 2.8 | 28.2 ± 3.9 | >50 | 1.57 ± 0.11 |
| ZR75.1 | 31.4 ± 3.2 | 28.6 ± 4.4 | >50 | >50 | >50 | 18.2 ± 2.2 | 29.6 ± 3.3 | >50 | 0.14 ± 0.02 |
The cellular effects of Hsp90 are mediated in large part by its physical association with several client proteins, which contribute to tumor cell survival, including CDK4, CDK6, AKT, c-Raf, EGFR, HER2/neu, c-Kit.34 In addition, the cytoprotective role of Hsp90 has been linked to its ability to modulate the conformation and activity of the antiapoptotic protein survivin.35 In this context, we examined whether the cytotoxic effect exerted by compounds 2, 4, 19 and 20 was due to the breakdown of multiple cell survival pathways as a consequence of destabilization of Hsp90 client proteins. Specifically, the exposure of MCF-7 and ZR75.1 cells to IC50 and IC80 of compounds 2, 4 and 19 resulted in a decrease in the expression levels of AKT, CDK4, CDK6, and survivin (see Fig. 6). The treatment did not modify the Hsp90 expression levels, whereas an increase of Hsp70 protein abundance was appreciable (see Fig. 6). By contrast, the cytotoxic effect exerted by compound 20 does not seem to rely on the interference with the activity of Hsp90. In fact, no alteration in the expression of Hsp90 client proteins was observed after treatment in both cell lines (see Fig. 6).
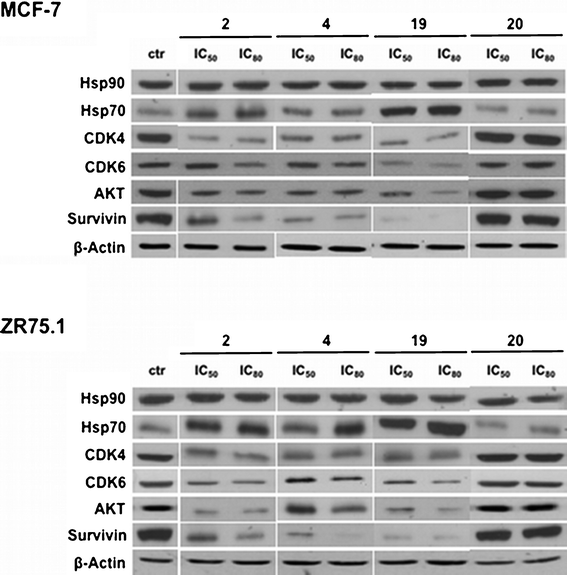 | ||
| Fig. 6 Effects of small molecules on Hsp90 chaperone function. Western blotting experiments showing the loss of Hsp90 client proteins in breast cancer cells treated with 1% DMSO (v/v) (control cells; ctr), or with compounds 2, 4, 19 or 20 (IC50 and IC80) for 72 h. | ||
On the basis of the above data, since some of the Hsp90 clients are key cell cycle regulators, the effect of Hsp90 inhibition on cell cycle phase distribution was assessed. Consistent with the inhibition of both CDK4 and CDK6, the exposure of asynchronously growing MCF-7 and ZR75.1 cells to compounds 2, 4 and 19 induced a marked change in the distribution of the cells in the different phases of the cell cycle (Fig. 7A). Specifically, flow cytometric analysis of propidium iodide-stained cells showed that a 72-h treatment with subcytotoxic, IC50, and IC80 concentrations of compounds 2, 4 and 19 resulted in a concentration-dependent accumulation of cells in G0/G1 phase (see Fig. 7A). In addition, chromatin condensation and DNA fragmentation, which are common features of apoptosis, were detected by fluorescence microscopy after staining with propidium iodide of breast cancer cells treated with small molecules. The percentage of cells with an apoptotic nuclear morphology, as determined on the overall cell population, increased in a concentration-dependent fashion after treatment with all compounds (see Fig. 7B).
 | ||
| Fig. 7 Effects of small molecules and 17-AAG on cell cycle phase distribution and induction of apoptosis. Cells were exposed for 72 h to 1% DMSO (v/v) (control cells; ctr) or to compounds 2, 4, 19 or 17-AAG (IC20, IC50 and IC80). (A) Cell-cycle phase distribution was assessed by flow cytometric analyses. The percentage of cells in G0/G1 (black), S (white) and G2/M (grey) phases are shown. Columns represent the mean of three independent experiments; SDs are always within ±5%. (B) Percentage of cells with an apoptotic morphology as assessed by fluorescence microscopy. Data represent the mean values ±SD of three independent experiments. **P < 0.001 and *P < 0.01, student's t test. | ||
Notably, the effects on cell cycle phase distribution and induction of apoptosis observed after treatment with compounds 2, 4 and 19 are very similar to those obtained after exposure of MCF-7 and ZR75.1 cells to 17-AAG (see Fig. 7).
Analysis of the molecular interactions established by the most promising hits with Hsp90
The interactions of active hits with Hsp90 were analyzed in details by generating structural models that take into account different conformations of the receptor. In particular, we focused on compounds 2 and 19 since compound 4 shows interesting similarities with the ATP and ADP molecules (see Fig. 3) and, therefore, its modes of interaction with the molecular chaperone can be easily anticipated. In fact, it is possible to observe that in compound 4 we have a cytosine unit, which can be considered a mimic for the adenine in ATP/ADP, and, above all, a ribose ring and a phosphate group, which have exact correspondences in the two nucleotides.As described in details in the “Material and Methods” section, the other two promising hits (compounds 2 and 19) were docked into the ATP binding site of all the Hsp90 conformations considered for the construction of our pharmacophore model. After each docking calculation we extracted the Hsp90-inhibitor complex associated with the lowest binding energy conformation and we have afterwards analyzed the corresponding intermolecular interactions. As we will show below, this protocol has allowed us to identify the most stabilizing interactions for the compounds under examination (frequency greater than or equal to five), taking into account also protein fluctuations. However, before analyzing the obtained results, it is worthwhile to point out that compounds 2 and 19 are constituted by “quasi-equivalent” units (see Fig. 8 and 9), which have led to degeneracy in the docking solutions. Therefore, in order to better rationalize the results of our calculations, we have grouped together the interactions established by “quasi-equivalent” functional groups. To accomplish this task we have previously defined the following groups for compound 2 (see Fig. 8): the aromatic amino groups –N(1)H2, –N(27)H2 and –N(37)H2 (from now on indicated as ARAM), the aromatic rings (AR), the sulfonyl groups –S(8)O2, –S(19)O2 and –S(29)O2 (SU) and the aliphatic amino groups –N(11)H and –N(17)H (ALAM). In analogous way, for compound 19 we have (see Fig. 9): the hydroxyl groups of the sugar-like units –O(1)H, –O(30)H, –O(36)H and –O(28)H (from now on indicated as OG), the oxygen heteroatoms of the sugar-like units O(7) and O(24) (OE), the sugar-like units (SLU) and the purine units (PU).
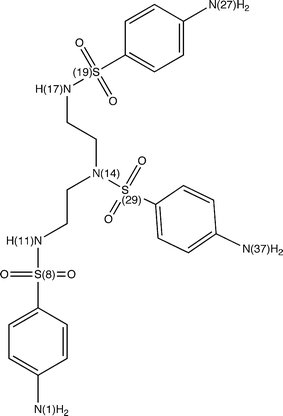 | ||
| Fig. 8 Compound 2 with labels to identify its “quasi-equivalent” units. | ||
 | ||
| Fig. 9 Compound 19 with labels to identify its “quasi-equivalent” units. | ||
Let us consider the interactions established by compound 2. From analysis of Tables 5 and 6 it is easy to see that the sulfonyl groups of the molecule act as hydrogen bond acceptors and form recurring interactions with Asn 37, Asn 92, Gly 121, Val 122, Gly 123, Phe 124 and Tyr 125, while the aromatic amino groups are hydrogen bonds donors and mainly interact with Asp 79 and Asn 92. It is interesting to note that, as discussed above, the interaction with Asp 79 is a recurring and fundamental hydrogen bond in many complexes between Hsp90 and known inhibitors,29–32,36 whereas the interactions (interaction) with Gly 123 and Phe 124 (Tyr 125) have (has) already been observed for geldanamycin (PU24FCl).29,31 Taking into account the hydrophobic contacts (see Table 7), the aromatic rings establish significant interactions with Ala 41, Ile 82, Met 84, Leu 93, Met 116, Phe 124, Val 136 and Leu 173, residues that are involved in hydrophobic contacts also with the already known inhibitors mentioned above.29–32 For instance, the aromatic ring face of radicicol forms extensive hydrophobic interactions with the side chains of Met 84, Leu 93, Phe 124, Thr171 and Leu 17330 while PU-H71 establishes hydrophobic contacts with Ala 41, Met 84 Phe 124 and Trp 148.31
| Hydrogen bond donor | hydrogen bond acceptor | Frequency |
|---|---|---|
| Asn 37 (side-chain) | SU | 8 |
| Asn 92 (side-chain) | SU | 7 |
| Gly 123 (backbone) | SU | 6 |
| Phe 124 (backbone) | SU | 6 |
| Tyr 125 (side-chain) | SU | 6 |
| Gly 121 (backbone) | SU | 5 |
| Val 122 (backbone) | SU | 5 |
| Phe 120 (backbone) | SU | 4 |
| Arg 345 (side-chain) | SU | 4 |
| Lys 102 (backbone) | SU | 3 |
| Gly 100 (backbone) | SU | 2 |
| Gln 119 (backbone) | SU | 2 |
| Hydrogen bond acceptor | Hydrogen bond donor | Frequency |
|---|---|---|
| Asp 79 (side-chain) | ARAM | 6 |
| Asn 92 (backbone) | ARAM | 5 |
| Thr 171 (side-chain) | ARAM | 4 |
| Asn 37 (side-chain) | ALAM | 3 |
| Leu 34 (backbone) | ARAM | 2 |
| Asp 40 (side-chain) | ALAM | 2 |
| Met 84 (side-chain) | ALAM | 2 |
| Gly 94 (backbone) | ARAM | 2 |
| Ser 99 (backbone) | ARAM | 2 |
| Ser 99 (side-chain) | ARAM | 2 |
| Met 116 (backbone) | ALAM | 2 |
| Ile 117 (backbone) | ARAM | 2 |
| Gln 119 (backbone) | ARAM | 2 |
| Hydrophobic residue | Inhibitor interacting unit | Frequency |
|---|---|---|
| Met 84 | AR | 12 |
| Leu 173 | AR | 11 |
| Ile 82 | AR | 9 |
| Phe 124 | AR | 9 |
| Ala 41 | AR | 8 |
| Leu 93 | AR | 7 |
| Met 116 | AR | 7 |
| Val 136 | AR | 6 |
| Ala 38 | AR | 4 |
| Leu 89 | AR | 4 |
| Ile 117 | AR | 3 |
| Leu 34 | AR | 2 |
| Ile 77 | AR | 2 |
| Ala 97 | AR | 2 |
| Ala 103 | AR | 2 |
| Met 116 | AR | 2 |
| Val 122 | AR | 2 |
| Leu 127 | AR | 2 |
In an analogous way, for compound 19 we have observed that the oxygen atoms of the hydroxyl groups act as hydrogen bond acceptors with Asn 37, Gly 83, Phe 120 and Gly 123, whereas the oxygen heteroatoms of the sugar-like units accept hydrogen bonds from the side chain of Gln 119 (see Table 8). Furthermore, also in this case the residue Asp 79 is crucial since it forms recurring hydrogen bond interactions with the hydroxyl groups of the molecule (see Table 9). Finally, considering the hydrophobic contacts we have a similar situation to the one observed for compound 2. In fact, the purine units of compound 19 mainly establish hydrophobic interactions with Ala 41, Met 84, Leu 93, Met 116 and Phe 124 (see Table 10).
| Hydrogen bond donor | Hydrogen bond acceptor | Frequency |
|---|---|---|
| Gly 83 (backbone) | OG | 6 |
| Asn 37 (side-chain) | OG | 5 |
| Gln 119 (side-chain) | OE | 5 |
| Phe 120 (backbone) | OG | 5 |
| Gly 123 (backbone) | OG | 5 |
| Gly 100 (backbone) | OG | 4 |
| Gly 121 (backbone) | OG | 4 |
| Val 122 (backbone) | OG | 4 |
| Thr 171 (side-chain) | OG | 4 |
| Arg 345 (side-chain) | OG | 4 |
| Arg 356 (side-chains) | OE | 4 |
| Ile 82 (backbone) | OG | 3 |
| Asn 37 (side-chain) | OE | 3 |
| Gln 119 (backbone) | OG | 3 |
| Gly 100 (backbone) | OE | 2 |
| Lys 102 (backbone) | OG | 2 |
| Lys 102 (backbone) | OE | 2 |
| Ala 103 (backbone) | OG | 2 |
| Phe 124 (backbone) | OG | 2 |
| Hydrogen bond acceptor | Hydrogen bond donor | Frequency |
|---|---|---|
| Asp 79 (side-chain) | OG | 10 |
| Asn 37 (side-chain) | OG | 4 |
| Lys 98 (backbone) | OG | 4 |
| Ser 99 (backbone) | OG | 3 |
| Ser 99 (backbone) | OG | 3 |
| Glu 33 (side-chain) | OG | 2 |
| Glu 88 (side-chain) | OG | 2 |
| Met 116 (backbone) | OG | 2 |
| Tyr 125 (side-chain) | OG | 2 |
| Thr 171 (side-chain) | OG | 2 |
| Hydrophobic residue | Inhibitor interacting unit | Frequency |
|---|---|---|
| Met 84 | PU | 9 |
| Leu 93 | PU | 6 |
| Phe 124 | PU | 6 |
| Ala 41 | SLU | 5 |
| Met 116 | PU | 5 |
| Ile 82 | SLU | 3 |
| Ala 38 | SLU | 2 |
| Ala 41 | PU | 2 |
| Ile 82 | PU | 2 |
| Met 116 | SLU | 2 |
| Leu 127 | PU | 2 |
| Leu 173 | PU | 2 |
Discussion
The main goal of this work consists in the proposal of a novel simple, rational in silico approach for the search of new possible small-molecule inhibitors taking into account the fundamental concepts of protein flexibility and conformational dynamics. In particular, the new strategy is based on the consideration that a ligand selectively binds one (or a subset) of the energetically similar protein conformers, stabilizing it (them) and inducing some specific protein conformations that cannot be predicted through a simple analysis of the corresponding unbound crystal structure. From an applicative point of view, considering the active protein–ligand complex, the proposed approach consists of a proper cluster analysis of a long-time scale all-atom Molecular Dynamics simulation to extract a minimal number of key configurations that take into account most of the structural fluctuations and, afterwards, in identifying the most recurring fundamental interactions between the protein and the ligand, interactions that allow to design a dynamic pharmacophore for the screening of small-molecule databases.In this manuscript the examined protein is the molecular chaperone Hsp90 that is generally viewed as a “druggable” target for rational cancer therapy due to (a) its role at the crossroads of multiple signalling pathways associated with cell proliferation and cell viability and (b) to its elevated ATPase activity in tumors as opposed to normal tissues.37 Exploiting our novel drug-discovery rational strategy, we have been able to identify new alternative N-terminal inhibitors explicitly including the flexibility and the conformational dynamics of the activated complex between Hsp90 and its natural ligand ATP. Importantly, the newly discovered Hsp90 inhibitors show interesting activities in blocking Hsp90 dependent cell pathways and interactions, as proved by the selective degradation of Hsp90 clients and the consequent cytotoxicity.
In this case, after selecting the seven most relevant conformational substates visited during the MD simulation of the complex, we have analyzed the occurrence of the intermolecular interactions between the ligand and the protein. In particular, we have observed that the intermolecular interactions that stabilize most the Hsp90–ATP complex are hydrogen bond-like interactions and, therefore, after identifying the hydrogen bond donors and acceptors in the ATP molecule, we have assigned them a weight proportional to the frequency with which they are involved in significant interactions in the extracted key configurations. This allowed us to design a dynamic pharmacophore model that implicitly takes into account the protein flexibility and that has been used to screen the small-molecule database NCI_3D.
A possible important limitation of our pharmacophore model is that it is mainly based mainly on mimicking hydrogen bond interactions. This may reverberate in the selection of molecules bearing charged groups, which is an undesirable feature in drug-like molecules. To alleviate this problem, we have included an additional level of control based on the application of the Lipinski rules.13 The virtual screening with this further filter returned 25 compounds, eight of which have been obtained from the National Cancer Institute and tested in a panel of human breast carcinoma cell lines. The logP values of all the 25 hits fulfil the Lipinski rule for the water–octanol partition coefficient13 and, above all, eight compounds respect the stricter Ghose rule33 as well. Three of the molecules passing the more stringent test are active against Hsp90, underlying the importance of including drug-like features in the second-stage analysis of the screening.
Furthermore, despite its limitations, the proposed pharmacophore model interestingly captures some of the binding determinants of the well-known inhibitors geldanamycin and PU24FCl. Indeed, both of these compounds are able to establish an extended network of direct hydrogen bonds with Hsp90. In contrast, our pharmacophore performs poorly with respect to other known inhibitors. However, it has to be noted that such compounds were designed and synthesized based on the careful analysis of X-ray structures, namely, using a totally different strategy compared to the one proposed here.
In fact, one of the aims of our effort was to consider explicitly the possible dynamic states of the receptor during the stage of the pharmacophore design. Including receptor flexibility may allow to take into account the formation/disappearance/change of pockets due to the conformational response of the protein to the ligand (ATP in our case). In turn, this enables the identification of the stereochemical properties of functional groups that should be present in a putative inhibitor to facilitate the optimal fitting to variations in the shape of the binding site. In this framework, the goal of our pharmacophore design was to select molecular entities that are different from known active compounds.
Four of the tested molecules (2, 4, 19 and 20) have shown an interesting cytotoxic effect and they have been subject to further experimental tests. In particular, exploiting the fact that the chaperone Hsp90 oversees the correct conformational development of a diverse ensemble of client proteins, we have investigated whether the cyotoxic effect exerted by the compounds relies on the interference with Hsp90 chaperone activity. In particular, we have observed that three compounds (2, 4 and 19) induce a decrease in the expression levels of Hsp90 client proteins for the cell lines MCF-7 and ZR75.1 and, furthermore, other tests have shown that the inhibition of some Hsp90 client proteins also results in a significant change in the distribution of the cells in the different phases of the cell cycle. In particular, the percentage of cells in apoptosis increases as the concentration of administrated compounds rises.
Finally, in order to gain an insight into possible structural determinants of the inhibitory/cellular activity of the most promising hits, we have studied their modes of interaction with Hsp90. To accomplish this task we have docked the active compounds into the ATP binding site of all the Hsp90 conformers used for the pharmacophore construction. Afterwards, from the statistical analysis of the intermolecular interactions in all the obtained Hsp90-inhibitor complexes, we have been able to identify the most recurring and stabilizing contacts and we have shown that they generally correspond to the ones already observed for a wide range of different known inhibitors. The fact that our active compounds reproduce the molecular recognition determinants of highly optimized drug candidates (e.g., hydrogen bonding similarities to geldanamycin and hydrophobic interactions similarities to the purine-based inhibitors and related derivatives) suggests that their function may be determined by similar mechanisms. In particular, we propose that the identified hits engage the ATP binding site, disrupting the Hsp90 ATPase activity that is necessary for the chaperone cycle.
At this point it is very important to stress again that the main goal of our investigations was not to dock Hsp90 inhibitors into the active site and to analyze the structures of the resulting complexes. The aim of our study was to identify the possible molecular determinants, and their dynamic evolution, required for binding to a certain pocket using the natural ligand as a template and to include these findings in the pharmacophore design stage. Afterwards, this explicit information on the protein conformational motions was used to search for novel structural entities that may interfere with the biological functions of the target.
We believe that the proposed strategy can be also extended to other targets and exploited to identify active compounds using information obtained directly from the atomic-resolution analysis of the dynamics of complex biomolecules. However, the “drug-like properties” of the obtained hits must be carefully examined before starting experimental tests through the application of proper filters, such as the Lipinski13 or Ghose33 rules. In fact, a pharmacophore such as the one proposed here may actually be biased towards selecting molecules with a number of charged groups which would make them fail as drug candidates. In this respect, improvements in the pharmacophore design process are clearly desirable.
In the application reported in this paper the activities of molecules are certainly still distant from the ones required for efficient pharmacological applications. Therefore, from this point onwards, we believe that classical drug design and medicinal chemistry efforts can be started to improve the activities and pharmacokinetic properties of the compounds.
In conclusion, considering the obtained results, it is worth pointing out again that the inclusion of protein flexibility is an important factor to be considered in the framework of rational drug discovery. This task can be accomplished simply considering a minimal number of key configurations that capture most of the structural fluctuations encountered in multi-nanosecond trajectories. The rate-limiting step in the whole procedure might be represented by the time required to run MD simulations. However, the fast pace in methodological developments and the continuous increase in computer power provide unprecedented chances to include atomic level descriptions of molecular motions in the drug discovery process.38–40 Therefore, we believe that our new technique can open the possibility to rationally expand the chemical space of the inhibitors for any other protein target involved in cancer development and it can help in discovering new possible uses for already known drugs.
Acknowledgements
A. G. and G. C. thank A. I. R. C. (Project IG. 11775), the PRIN project 2008K37RHP_002 and the project “Ministero della Salute Progetto Giovani Ricercatori” for financial support. We acknowledge the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute (NCI/DTP, dtp.nci.nih.gov) for providing the compounds used in our assays.References
- H. A. Carlson, K. M. Masukawa and J. A. McCammon, J. Phys. Chem. A, 1999, 103, 10213 CrossRef CAS.
- H. A. Carlson, K. M. Masukawa, K. Rubins, F. D. Bushman, W. L. Jorgensen, R. D. Lins, J. M. Briggs and J. A. McCammon, J. Med. Chem., 2000, 43, 2100 CrossRef CAS.
- H. A. Carlson, Curr. Opin. Chem. Biol., 2002, 6, 447 CrossRef CAS.
- K. L. Meagher and H. A. Carlson, J. Am. Chem. Soc., 2004, 126, 13276 CrossRef CAS.
- K. L. Damn and H. A. Carlson, J. Am. Chem. Soc., 2007, 129, 8225 CrossRef.
- A. L. Bowman, Z. Nikolovska-Coleska, H. Zhong, S. Wang and H. A. Carlson, J. Am. Chem. Soc., 2007, 129, 12809 CrossRef CAS.
- M. G. Lerner, A. L. Bowman and H. A. Carlson, J. Chem. Inf. Model., 2007, 47, 2358 CrossRef CAS.
- P. Workman, Curr. Cancer Drug Targets, 2003, 3, 297 CrossRef CAS.
- M. M. U. Ali, S. M. Roe, C. K. Vaughan, P. Meyer, B. Panaretou, P. W. Piper, C. Prodromou and L. H. Pearl, Nature, 2006, 440, 1013 CrossRef CAS.
- S. Frey, A. Leskovar, J. Reinstein and K. Buchner, J. Biol. Chem., 2007, 282, 35612 CrossRef CAS.
- M. Mickler, M. Hessling, C. Ratzke, J. Buchner and T. Hugel, Nat. Struct. Mol. Biol., 2009, 16, 281 CAS.
- J. Trepel, M. Mollapour, G. Giaccone and L. Neckers, Nat. Rev. Cancer, 2010, 10, 537 CrossRef CAS.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3 CrossRef CAS.
- G. Morra, M. A. C. Neves, C. J. Plescia, S. Tsustsumi, L. Neckers, G. Verkhivker, D. C. Altieri and G. Colombo, J. Chem. Theory Comput., 2010, 6, 2978 CrossRef CAS.
- D. Van der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701 CrossRef CAS.
- W. R. P. Scott, P. H. Hunenberger, I. G. Tironi, A. E. Mark, S. R. Billeter, J. Fennen, A. E. Torda, T. Huber, P. Kruger and W. F. van Gunsteren, J. Phys. Chem. A, 1999, 103, 3596 CrossRef CAS.
- H. J. C. Berendsen, J. R. Grigera and P. R. Straatsma, J. Phys. Chem., 1987, 91, 6269 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463 CrossRef CAS.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. Di Nola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684 CrossRef CAS.
- F. Pontiggia, A. Zen and C. Micheletti, Biophys. J., 2008, 95, 5901 CrossRef CAS.
- L. Kaufman, P. J. Rousseuw, in Finding Groups in Data: An Introduction to Cluster Analysis, Wiley, New York, 2005 Search PubMed.
- Discovery Studio 2.5, Accelrys, USA, 2009.
- Instant J Chem, 2.0.0 , 2006, ChemAxon (http://chemaxon.com).
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107 CrossRef CAS.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785 CrossRef CAS.
- C. E. Stebbins, A. A. Russo, C. Schneider, N. Rosen, F. U. Hartl and N. P. Pavletich, Cell, 1997, 89, 239 CrossRef CAS.
- S. M. Roe, C. Prodromou, R. O'Brien, J. E. Ladbury, P. W. Piper and L. H. Pearl, J. Med. Chem., 1999, 42, 260 CrossRef CAS.
- R. M. Immormino, Y. Kang, G. Chiosis and D. T. Gewirth, J. Med. Chem., 2006, 49, 4953 CrossRef CAS.
- T. E. Barta, J. M. Veal, J. W. Rice, J. M. Partridge, R. P. Fadden, W. Ma, M. Jenks, L. Geng, G. J. Hanson, K. H. Huang, A. F. Barabasz, B. E. Foley, J. Otto and S. E. Hall, Bioorg. Med. Chem. Lett., 2008, 18, 3517 CrossRef CAS.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Comb. Chem., 1999, 1, 55 CrossRef CAS.
- M. Taipale, D. F. Jarosz and S. Lindquist, Nat. Rev. Mol. Cell Biol., 2010, 11, 515 CrossRef CAS.
- P. Fortugno, E. Beltrami, J. Plescia, J. Fontana, D. Pradhan, P. C. Marchisio, W. C. Sessa and D. C. Altieri, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13791 CrossRef CAS.
- M. Meli, M. Pennati, M. Curto, M. G. Daidone, J. Plescia, S. Toba, D. C. Altieri, N. Zaffaroni and G. Colombo, J. Med. Chem., 2006, 49, 7721 CrossRef CAS.
- A. Kamal, L. Thao, J. Sensintaffar, L. Zhang, M. F. Boehm, L. C. Fritz and F. J. Burrows, Nature, 2003, 425, 407 CrossRef CAS.
- D. E. Shaw, P. Maragakis, K. Lindorff-Larsen, S. Piana, R. O. Dror, M. P. Eastwood, J. A. Bank, J. M. Jumper, J. K. Salmon, Y. B. Shan and W. Wiggers, Science, 2010, 330, 341 CrossRef CAS.
- Y. B. Shan, E. T. Kim, M. P. Eastwood, R. O. Dror, M. A. Seeliger and D. E. Shaw, J. Am. Chem. Soc., 2011, 133, 9181 CrossRef CAS.
- R. O. Dror, A. C. Pan, D. H. Arlow, D.W. Borhami, P. Maragakis, Y. B. Shan, H. Xu and D. E. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13118 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |