Catalytic versus stoichiometric dehydrocoupling using main group metals
Robert J.
Less
,
Rebecca L.
Melen
and
Dominic S.
Wright
*
Chemistry Department, Cambridge University, Lensfield Road, Cambridge, CB2 1EW. E-mail: dsw1000@cam.ac.uk; Fax: 0044 1223 336362; Tel: 0044 1223 763122
First published on 9th January 2012
Abstract
A primary factor influencing catalyticversus stoichiometric behaviour of molecular main group species in homogeneous dehydrocoupling reactions is the redox stability of the metal centre. Thus, only in the case of redox-stable metals has catalytic behaviour so far been observed, through genuinely hydrogenic coupling (E–H + E′–H → E–E′ + H2), whereas for redox-unstable metals oxidative dehydrocoupling is seen (E–H + E′–H → E–E′ + 2H+ + 2e). The mechanisms of catalytic P–H/P–H and B–H/N–H dehydrocoupling involving main group systems are closely related to d0 transition metal counterparts and produce a similar range of products, although the main group systems reported so far are not as active as the most active transition metal catalysts.
 Robert J. Less | Robert J. Less was born in London in 1975. He undertook his first degree in Cambridge (1993–1996), moving on to a Ph.D. at Cambridge under Dr J. M. Rawson (1996–1999). He then went on to do postdoctoral research with Prof. P. R. Raithby (Bath) and Dr R. P. Davies (Imperial College, London) before returning to Cambridge in 2007. He is currently a Leverhulme postdoctoral fellow in the Wright group. |
 Rebecca L. Melen | Rebecca L. Melen was born in Nottingham in 1985. She undertook her first degree in Cambridge (2004–2008) and is currently a Ph.D. student in the Wright group working on phosphane chemistry and main group metal mediated B–H/N–H dehydrocoupling. |
 Dominic S. Wright | Dominic S. Wright obtained his first degree at Strathclyde University (1982–1986) before moving to Cambridge University where he did his Ph.D. under the late Dr Ron Snaith (1986–1989). After a research fellowship at Gonville and Caius College Cambridge (1989–1991), he was appointed to a lectureship at Cambridge and promoted to Reader in Inorganic Chemistry in 2002 and promoted to a personal chair in Inorganic Chemistry in 2010. He is the author of around 230 academic papers. |
Introduction
Transition metal catalysts have enjoyed a pre-eminent place in the homogeneous and heterogeneous catalysis of a large number of technologically important reactions. According to conventional wisdom this activity is fundamentally associated with the use of valence d-orbitals in bonding, which results in the special ability of these metals to activate precursor molecules (via synergic bonding). The presence of accessible d-orbitals gives transition metals the ability to change oxidation state readily and results in generally greater covalency within metal–ligand bonds than occurs in main group counterparts.1 This feature, combined with labile ligand coordination chemistry, underpins the use of transition metal compounds in an extremely broad range of catalytic and non-catalytic bond-forming reactions. But is the behaviour of transition metal complexes always so distinctly different from main group compounds? This important question has been highlighted by Power in regard to recent observations that the chemistry of the heavier main group elements ‘more closely resembles transition metals in their coordination and reactions with small molecules (like H2, CO, NH3 and C2H4) than that of the lighter main group congeners’.2Studies in the past decade have uncovered general patterns of reactivity which span the perceived divide between main group and transition metal chemistry, by showing that d0 main group metals can also be highly active in catalytic bond-forming reactions.3–7 There is therefore the potential to replace a number of key transition metal mediated reactions with cheaper common main group metals. There is currently world-wide interest in the synthetic applications of a variety of transition metal complexes in catalytic and non-catalytic main group element bond-forming reactions, via homo- or hetero-dehydrocoupling of E–H bonds (eqn (1)).8 This new field has been termed inorganometallic because of its relationship to the now ubiquitous use of transition metal species in organic transformations (organometallic),9 and has major future applications in molecular and polymer synthesis as well as hydrogen storage.10
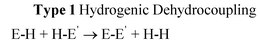 | (1) |
Pertaining to the field of future energy resource, an emerging area of interest is the use of amine–boranes, R2NHBH3, as light-weight materials for the chemical storage of H2 (eqn (2)).10,11 One of the prerequisites for the applications of these species (e.g., in fuel cells) is the ability to release H2 rapidly at low temperatures. A particularly important case is ammonia borane (NH3BH3) which is a promising hydrogen-storage material (containing 19.6% by weight hydrogen) but which decomposes at temperatures in excess of 120 °C.10 Hence, a great deal of effort has gone into the development of transition metal catalysts for this dehydrogenation step. However, the majority of these species are based on precious metals, such as Rh, Ru and Pd.10 There would be significant advantages in using far cheaper and more plentiful main group metals in this setting.12
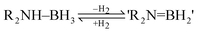 | (2) |
In a previous review,3a we focused on the stoichiometric dehydrocoupling of P–H bonds into P–P bonds (eqn (1), E = E′ = P) involving main group metal reagents. Here we explore the latest developments in the area of dehydrocoupling reactions involving main group metal catalysis, with the emphasis on the heteroatomic dehydrocoupling of B–H and N–H bonds (eqn (1), E = B, E′ = N). To the best of our knowledge this area has not previously been reviewed. The key issues addressed are (i) the mechanism and kinetics of dehydrocoupling involving main group metals, (ii) its comparison with transition metal systems, and (iii) the synthetic potential of main group dehydrocoupling reactions.
Stoichiometric vs.catalytic dehydrocoupling in P–H/P–H dehydrocoupling
Transition metal catalysed P–H/P–H or B–H/N–H dehydrocoupling can be seen to involve two overall types of reaction pathway. For organometallic catalysts involving partial dn (n ≠ 0) configurations, the course of the catalytic pathway takes place at least in part through oxidative addition and reductive elimination cycles, although the actual mechanisms are in many cases more complicated, with different products resulting from the use of different catalysts and phosphine and amine–borane precursors.8,10 The general features of dncatalysts of this class can be illustrated by a number of studies in this area, but a simple example investigated by Tilley and coworkers is the catalytic dehydrocoupling of primary phosphines (RPH2) to diphosphanes [RP(H)P(H)R] (2) by the RhI catalyst [(dppe)Rh(η3–CH2C6H5)] (1) [dppe = Ph2P(CH2)PPh2], in involving a RhI/RhIII cycle (Scheme 1).13 A change in the oxidation state of the transition metal centre during the course of the catalytic cycle also occurs for the dehydrocoupling of amine–boranes (R2NHBH3) with a range of low-oxidation state and/or later transition metal catalysts. The mechanism of activation, although still uncertain in respect to the individual steps in the sequence, is thought to be dominated by the cycle shown in Scheme 1. An example of this that has been particularly thoroughly studied is the B–H/N–H dehydrocoupling using the 12eRhI cation [(PPh3)2Rh]+ by Weller and coworkers (Scheme 2).14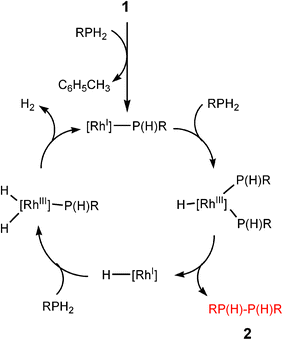 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
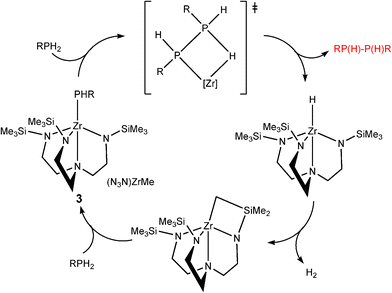
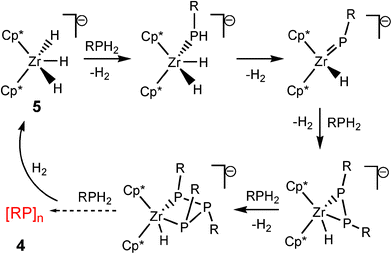
The second class of transition metal species contain d0 metal centres. These are distinguished from the dncatalytic systems in that the catalytic cycles do not involve a change in the oxidation state of the metal.8 Good examples of this can be seen in studies of the dehydrocoupling of phoshines by the groups of Waterman15 and Stephan involving TiIV and ZrIV.16 For example, the catalytic formation of diphosphanes RP(H)P(H)R (2) with (N3N)ZrMe (3) [N3N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(CH2CH2NSiMe3)32−] from primary phosphines (RPH2) is illustrated in Scheme 3.15 The dependence of the product on the catalytic system used is emphasised by the fact that, in contrast to the previous reaction, primary phosphines are converted into cyclic phosphanes ([RP]n) 4 using the anion [Cp*2ZrH3]− (5) as the catalyst (Scheme 4).16 Recent studies have also extended the applications of Group 4 d0 precatalysts like Ti(NMe2)4 in the dehydrogenation of amine–boranes.17
N(CH2CH2NSiMe3)32−] from primary phosphines (RPH2) is illustrated in Scheme 3.15 The dependence of the product on the catalytic system used is emphasised by the fact that, in contrast to the previous reaction, primary phosphines are converted into cyclic phosphanes ([RP]n) 4 using the anion [Cp*2ZrH3]− (5) as the catalyst (Scheme 4).16 Recent studies have also extended the applications of Group 4 d0 precatalysts like Ti(NMe2)4 in the dehydrogenation of amine–boranes.17
 | ||
| Scheme 3 | ||
 | ||
| Scheme 4 | ||
The significance of the activity of d0 transition metals in catalytic dehydrocoupling reactions, in respect to the focus of this review, is the strong relationship of active species like [(N3N)ZrMe] (3) and [Cp*2ZrH3]− (5) to many main group (s- and p-block) metal compounds. Thus, these catalysts have a high degree of ionic character, are highly basic and there is minimal involvement of valence d-orbitals in bonding (which are contracted in such high oxidation states). It is perhaps no surprise then that simple main group bases like metal dialkyl- and silyl-amides, and organometallics (see later, Fig. 3) have been found to be active in the dehydrocoupling of phosphines and amine–boranes. But what are the conditions for catalytic activity for main group bases of this type?
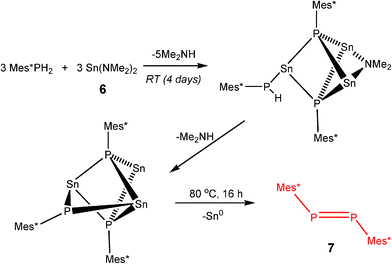
From extensive investigation of the reactivity of a range of p-block dimethylamido base, M(NMe2)n (n = 2, M = Sn; n = 3, M = As, Sb, Bi)3a it is clear that although these species are highly active in the dehydrocoupling of phosphines, these reactions are nonetheless stoichiometric. This is because this is not the same type of hydrogenic coupling that is found for transition metal catalytic systems (Type 1, eqn (1)) but rather oxidative coupling (Type 2, eqn (3)). This is a consequence of the combination of the basic character of these reagents (i.e., –2H+) and their redox instability (i.e., –2e) under the reaction conditions. Thus the net result is the formation either of metals (M0) or Zintl ions resulting from metal reduction from the noxidation state.3a,b,18 A recent example of this is seen in the dehydrocoupling of Mes*PH2 (Mes* = 2,3,5–tBu3C6H4) with Sn(NMe2)2 (6) (Scheme 5), giving Sn0 and the P=P bonded product Mes*P=PMes* (7).19 The reduction of M in this type of reaction does not appear to be reversible and hence this can be seen as the primary reason for the stoichiometric (rather than catalytic) outcome of the reaction. These observations highlight one potential difference between transition metal catalysed dehydrocoupling systems and those of the main group, that redox stability of the metals (M) is important to the maintenance of catalytic behaviour for main group reagents, and that there is currently no counterpart to the dncatalytic situation that occurs for transition metals.
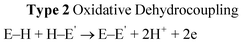 | (3) |
 | ||
| Scheme 5 | ||
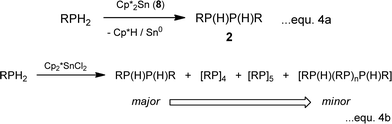
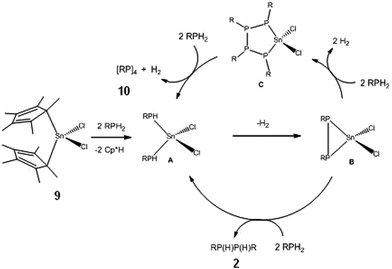
Further recent studies reiterate these conclusions. Firstly, it is found that [Cp*2SnII] (8) behaves in much the same way as Sn(NMe2)2 (6) (as a redox-active base) and functions as a stoichiometric reagent in the coupling of primary phosphines into diphosphanes (2) (eqn. (4a); cf. the catalytic behaviour in Scheme 1). However, switching to [Cp*2SnIVCl2] (9) as the reagent produces at least substoichiometric behaviour with Turn Over Numbers (TON) of up to 8 (at 60 °C, 4 days) which are not dissimilar to those found for the ZrIV catalysts discussed above (with TON = 11–20,8,15 at 90–120 °C, 1–9 days) (eqn. (4b)). This SnIV-mediated reaction is apparently hydrogenic (Type 1) and involves P–H activation by the C–Sn bonds, as suggested by the formation of Cp*H during the initial stage. A tentative mechanism is outlined in Scheme 6. The product is a mixture of the diphosphanes [RP(H)P(H)R] (2), the cyclic tetraphosphane [RP]4 (10), the pentaphosphane [RP]5 (11) and higher-chain oligomers [RP(H)(RP)nP(H)R] (n ≥ 1) (12). This range of products is similar to that obtained in the reactions of using [(N3N)ZrR], mentioned previously, as is the dependence of product distribution on the nature of the R-group.15
 | ||
| Scheme 6 | ||
In situ 31P NMR spectroscopic studies, combined with fractional crystallisation of the products produced over the course of the reaction, support the view that the low TON found for Cp*2SnCl2 (9) is due to rapid termination of the catalytic cycle via the irreversible reduction of SnIV to SnII. Relevant to the lower activity of SnIIversusSnIV is the recent observation that the catalytic activity of Group 2 vs.Group 3 amides in dehydrocoupling of amine–boranes decreases markedly with decreased Lewis acidity of the metal.5b In the dehydrocoupling of FcPH2 (Fc = ferrocenyl) with Cp*2SnCl2 the first product observed is the diphosphane [FcP(H)P(H)Fc] (13), then the tetraphosphane [FcP]4 (14) (Fig. 1a), next the pentaphosphane [FcP]5 (15) (Fig. 1b) and only at the end of the reaction (after 4 days) is the presumed SnII termination product [(FcPPFc)SnCl2]2 obtained (16) (Fig. 1c). The most likely source of SnII is from reductive elimination of ‘[RP=PR]’ or [RP]4 from intermediates like B and C in Scheme 6.
![a) the tetraphosphane [FcP]4 (14), the pentaphosphane [FcP]5(15) and c) the presumed SnII termination product (16).](/image/article/2012/RA/c2ra00882c/c2ra00882c-f1.gif) | ||
| Fig. 1 a) the tetraphosphane [FcP]4 (14), the pentaphosphane [FcP]5(15) and c) the presumed SnII termination product (16). | ||
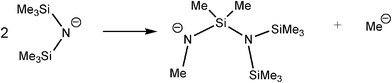
The lower redox stability of GaIII intermediates compared to AlIII counterparts, especially hydrides,20 also has the general effect of decreasing the catalytic activity of GaIII amides in B–H/N–H dehydrocoupling compared to AlIII counterparts (see later discussion).6 A dramatic example of this has been seen recently in the reaction of the GaIII base [Ga{N(SiMe3)2}3] (17) with ammonia–borane (NH3BH3).6b This reaction is stoichiometric (Type 2) rather than catalytic and gives the unusual, conjugated imido–borane [B{(NHBH)N(SiMe3)Si(Me2)N(SiMe3)2}3] (18) (Fig. 2) in which a combination of B–N bond formation and N–Si bond cleavage of the N(SiMe3)2 groups occurs (Scheme 7). This result can be traced to the powerful reducing effect of the finely-divided Ga metal decomposition product.21
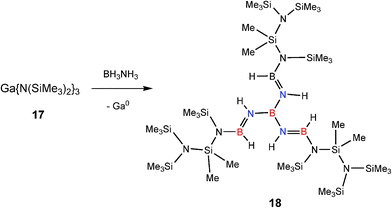 | ||
| Fig. 2 Structure of the conjugated imido borane generated from stoichiometric dehydrocoupling of NH3BH3 with Ga {N(SiMe3)2}3 (17). | ||
 | ||
| Scheme 7 | ||
Main group metal catalysed dehydrocoupling of amine–boranes
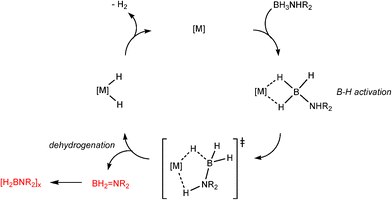
Developments in the area of catalytic dehydrocoupling of amine boranes using main group elements have proceeded rapidly in the past three years. It is perhaps no surprise, bearing in mind the previous discussion of catalyticvs. stoichiometric behaviour, that the realms of catalysis of amine boranes has so far been restricted to redox-stable alkaline earth, rare earth metals, and aluminium. The most likely reason for this is the redox stability of the metals involved and the consequent stability of metal hydride intermediates in these systems, which function as the active catalysts.The first evidence that molecular main group reagents could be active in B–N bond formation was obtained by Hill and coworkers who showed that the σ-bond metathesis reaction of β-diketiminato calcium amide complex 19 with the secondary organoborane 9-BBN (20) (9-borobicyclo[3,3,1]nonane) results in the B–N bonded product 21 and the calcium borohydride hydride complex 22 (Scheme 8).22 Since then a range of reagents have been investigated in stoichiometric and catalytic dehydrocoupling of amine boranes (Fig. 3).
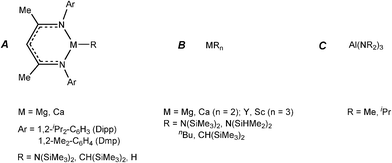 | ||
| Fig. 3 Main group reagents which have been employed in stoichiometric and catalytic studies with ammonia borane and amine–boranes. | ||
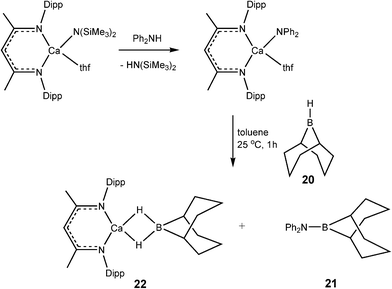 | ||
| Scheme 8 | ||
Later in situNMR spectroscopic and structural studies of the stoichiometric reactions of the β-diketiminato calcium and magnesium complexes (of Type A, Fig. 3), and silylamide and organometallic complexes (of Type B, Fig. 3) with ammonia–borane (NH3BH3),4a and a range of amine–boranes [R1R2NHBH3, R1 = H, R2 = Me, iPr, Dipp;[4bc,e]; R1 = R2 = Me or iPr]4d,f,g by the groups of Harder and Hill were able to identify key intermediates in the catalytic reactions. Some important representatives which were structurally characterised are shown in Fig. 4. For consistency, all of the species shown possess β-diketiminato ligands and result from reactions of type A reagents, however, similar types of intermediates have been uncovered using Type B reagents of Group 2 (Mg, Ca, Sr)4 and also Group 3 (Sc, Y).5
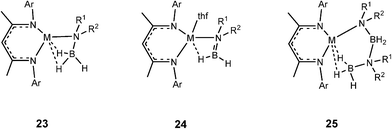 | ||
| Fig. 4 Key types of intermediates that have been structurally characterised. | ||
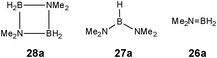
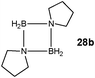
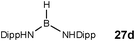
Although there is still some debate about precise mechanistic details,4c these studies lead Hill to suggest a potentially general mechanism for the catalytic cycle involved in the formation of the range of B–N products (Scheme 9).4d These products are highlighted in red in Scheme 9 and their formation depends primarily on the amine–borane used, and also to some extent on the kinetics of individual steps (particularly β–BHvs δ–BH elimination, producing the alternative products 27 and 28). For example, using the less sterically bulky Me2NHBH3 the products are (Me2N)2BH (27a), [Me2NBH2]2 (28a) and traces of Me2NBH2 (26a) (using cycles 1 and 2),4d,f,g,5 using more bulky iPr2NHBH3 the unsaturated imido–boraneiPr2NBH2 (26c) is produced (using only cycle 1),4f and using DippNH2BH3 gives (DippNH)2BH (27d) (cycles 1 and 2) (see Table 1).4c Similar products are generated using the same amine–boranes in a variety of transition metal catalysed reactions.10 The important observation that the greater the charge density of the Group 2 or 3 metals involved, the greater the catalytic activity has been ascribed to the effect of polarisation on the rate of σ-metathesis (β–BH and δ–B–H elimination) and on insertion of unsubstituted intermediates (like that in the proposed transition state 29, Scheme 9).4d,5
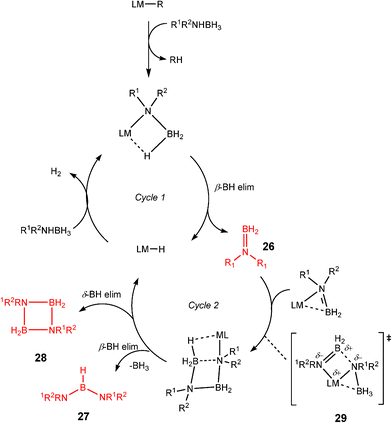 | ||
| Scheme 9 | ||
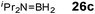
In common with transition metal catalytic systems (e.g., like that shown in Scheme 2) metal hydrides are proposed as key intermediates in these dehydrocoupling reactions. Although definitive evidence has been understandably difficult to obtain from in situNMR spectroscopic studies so far,4f strong support for this proposition, albeit indirect, was obtained from the isolation of the Zn hydride [CH{![[horiz bar, triple dot above]](https://www.rsc.org/images/entities/char_e0f1.gif) C(Me)N(Dipp)}2ZnH] (31) in high yield from the stoichiometric reaction of the chloride precursor 30 with the potassium salt K[iPrNHBH3] (Scheme 10).23
C(Me)N(Dipp)}2ZnH] (31) in high yield from the stoichiometric reaction of the chloride precursor 30 with the potassium salt K[iPrNHBH3] (Scheme 10).23
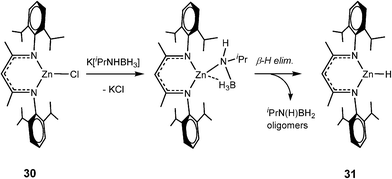 | ||
| Scheme 10 | ||
The mechanistic details found for the catalytic dehydrocoupling of AlIII amide bases Al(NR2)3 (Type C) with amine–boranes are similar to those shown in Scheme 9 for early main group (Type A and B) precatalysts.6 This situation is, however, complicated by the non-innocent involvement of the ligands. Firstly, the Me2N−groups of the less sterically encumbered Al(NMe2)3 (i.e., Me2N = L, in Scheme 9) can exchange with the R1R2NH−groups of the amine–borane (eqn (5)), leading to a complicated mixture of intermediates present in the catalytic reactions. The tendency for this reaction to occur depends on the amine–borane, R1R2NHBH3, so that (for example) comparatively little exchange of Me2N− and tBuNH− occurs for tBuNH2BH3 whereas a significant amount of exchange is observed for iPr2NHBH3.6b Recently, it has been suggested that in at least some cases this type of ligand exchange reaction may play a part in the overall catalytic cycle.24 Secondly, the R2N−groups of Al(NR2)3 precatalysts (Type C) can be intimately involved in generating the active catalyst species. This is seen in studies of the catalytic reaction of Al(NMe2)3 with Me2NHBH3 (Scheme 11) in which the active species is the dihydride 33, generated either by base-assisted β-hydride transfer to AlIII (as depicted) or by β–BH elimination followed by addition of Me2N− to a metal-bonded, unsaturated Me2NBH2. Recent evidence has suggested that reactions involving early main group Type B precatalysts may not be as simple as initially proposed in Scheme 9, and that here also the same type of addition of previously-assumed innocent ligands to unsaturated BN intermediates can occur.4g
| R1R2NHBH3 + Al(NMe2)3 → Me2NHBH3 + (R1R2N)Al(NMe2)2 | (5) |
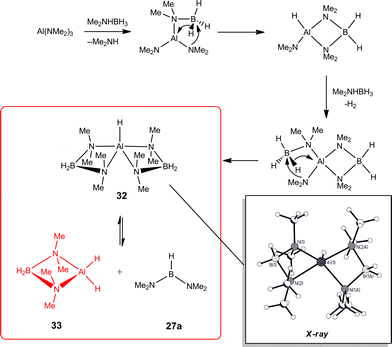 | ||
| Scheme 11 | ||
Once the catalyst 33 is established, the final B–N product [Me2NBH2]2 (28a) (Table 1) appears to be generated via a similar set of reactions to Cycle 2 in Scheme 9, involving δ–BH elimination. The big difference, however, is that the minor product in this reaction (Me2N)2BH (27a) (Table 1) is not formed by a β-hydride elimination within this cycle but only in the initiation stage during the formation of the catalyst 33via dissociation of 32 (Scheme 11). The absence of free Me2NBH2 (26a) (Table 1) as even a transient product in this reaction supports the view that the catalytic mechanism involved in this case is entirely metal-based.
An extremely long induction period of up to 30 min occurs in the case of the catalytic reaction of (iPr2N)3Al with iPr2NHBH3 as a result of the slow formation of the apparently active dihydride species [H2Al(μ-NiPr2)]2 (34) (Scheme 12), which is structurally similar to the catalytic species 33 involved in the reaction between Al(NMe2)3 and Me2NHBH3.6b In this case, termination of the catalytic cycle (which is similar to Cycle 1 in Scheme 9) apparently occurs by formation of an inactive BH4− complex (35) which only appears at the end of the reaction. This is closely related to the deactivation of the dihydride pincer catalyst [(POCOP)IrH2] [POCOP = η1-1,3-(OPtBu2)2C6H3] with BH3 in the dehydrogenation reaction with ammonia borane, NH3BH3.25 However, an alternative explanation for the final formation of a BH4− complex is that this is an additional catalytic species or a catalyst rest state.4c
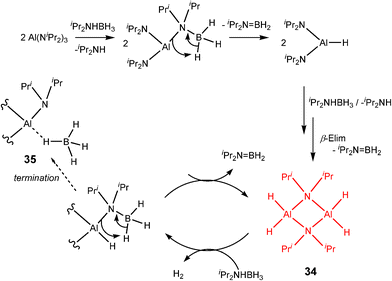 | ||
| Scheme 12 | ||
The reaction of Al(NMe2)3 with tBuNH2BH3 is of particular interest in respect to comparison with transition metal catalysis. This reaction gives a mixture of the trimeric borazane [BH2N(H)tBu]3 (36), as well as the borazine [BHNtBu]3 (37) and unidentified polymeric B–N compounds (Scheme 13).6b The apparent intermediacy of the borazane 36 in this reaction is similar to the dehydrocoupling reactions of a range of amine–boranes [RNH2BH3] (R = H, Me, Ph) with the transition metal catalyst [Rh(1,5-cod)(μ-Cl)]2.26 However, in the case of the RhI-catalysed reactions the rate of formation of the borazanes is much faster than that of the subsequent dehydrogenation to the borazines, resulting in slow build up in the borazane with time before it is slowly consumed to give the borazine. This is the opposite to the situation found in the AllII system in which the borazane and borazine are formed together as the reaction proceeds.
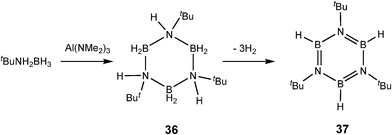 | ||
| Scheme 13 | ||
Comparison of rate data, transition metal vs. main group metal
A major obstacle to accurate comparison of the activity of main group and transition metal catalysed reactions in the area of dehydrocoupling is the lack of reported quantitative data like TON50 (Turnover Number at 50% conversion) and TOF (Turnover Frequency), and the broad range of solvents and conditions that have been employed, which may differ significantly from one reaction to another. More often than not the activity quoted in the literature is based purely on a semi-quantitative expression of a certain percentage conversion in a particular time. Table 2 lists all of the data currently available using main group precatalysts and converts this into approximate TON50 and TOF values so that some broad comparison can be made. The main conclusion which can be drawn is that, although not anywhere near as active as the most active transition metal dehydrocoupling catalysts recently introduced (with TON50ca. 5000 and TOF 6000 h−1),14,27 main group catalysts are comparable in activity with a broad range of other transition metal catalysts used to effect the same transformations. Thus the range of activities previously reported for the majority of dehydrocoupling reactions of Me2NHBH3, iPr2NHBH3 and tBuNH2BH3 involving transition metals (where data is available, from Table 2 reference 10a) correspond to TON50 = 4–100 and TOF = 0.1–2.8 h−1, which compares to TON50 = 5–100 and TOF = 0.1–30 h−1 for main group catalysis.| Substrate | Catalyst | % loading | Product | Approx. % conversion | Conditions | TON50 | TOF | Ref. |
|---|---|---|---|---|---|---|---|---|
| Me2NHBH3 | Mg[CH(SiMe3)2]2(thf)2 | 5 mol (%) | [Me2NBH2]2 | 100% | 60 °C, 72 h, C6D6 | 10 | 0.28 | 4d |
| Me2NHBH3 | Y[N(SiMe3)2]3 | 3 mol (%) | [Me2NBH2]2 | 86% | 60 °C, 12 h, C6D6 | 17 | 2.39 | 5 |
| Me2NHBH3 | Y[N(SiMe3)2]3 | 3 mol (%) | [Me2NBH2]2 | 4% | 25 °C, 0.25 h, C6D6 | 17 | 5.33 | 5 |
| Me2NHBH3 | Sc[N(SiMe3)2]3(thf)2 | 3 mol (%) | [Me2NBH2]2 | 10% | 25 °C, 1 h, C6D6 | 17 | 3.33 | 5 |
| Me2NHBH3 | Sc[N(SiMe3)2]3(thf)2 | 3 mol (%) | [Me2NBH2]2 | 90% | 60 °C, 1 h, C6D6 | 17 | 30.00 | 5 |
| Me2NHBH3 | Al(NMe2)3 | 8 mol (%) | [Me2NBH2]2 | 80% | 25 °C, 120 h, C7D8 | 6 | 0.08 | 6b |
| Me2NHBH3 | Al(NMe2)3 | 5 mol (%) | [Me2NBH2]2 | 100% | 50 °C, 48 h, C7D8 | 10 | 0.42 | 6b |
| i Pr2NHBH3 | Al(NiPr2)3 | 2 mol (%) | i Pr2NBH2 | 100% | 20 °C, 2 h, C7D8 | 25 | 25.00 | 6b |
| i Pr2NHBH3 | Al(NiPr2)3 | 10 mol (%) | i Pr2NBH2 | 100% | 60 °C, 2 h, C6D6 | 5 | 5.00 | 6b |
| i Pr2NHBH3 | [H2Al(μ-NiPr2)]2 | 0.5 mol (%) | i Pr2NBH2 | 50% | 20 °C, 96 h, C7D8 | 100 | 1.04 | 6b |
| i Pr2NHBH3 | Mg[N(SiMe3)2]2 | 5 mol (%) | i Pr2NBH2 | 100% | 25 °C, 1 h, C6D6 | 10 | 20.00 | 4f |
| t BuNH2BH3 | Al(NMe2)3 | 3 mol (%) | [tBuNBH]3 | 13% | 20 °C, 96 h, C6D6 | 17 | 0.05 | 6b |
Conclusions and future prospects
From the survey of the reactivity of main group metals in catalysis in this area it can be concluded that (i) the catalytic activity of homogeneous molecular main group metal catalysts is very dependent on the redox stability of the metal and that the course of dehydrocoupling reactions [whether dehydrogenic (catalytic) or stoichiometric (oxidative)] is determined by this redox stability, (ii) that the mechanisms of dehydrocoupling of phosphines (RPH2) and amine boranes (R1R2NHBH3) with d0 transition metals and main group metals are surprisingly similar and involve related intermediates, and (iii) that although the activity of main group catalysts is far lower than that of the most active transition metal systems, the activity is similar to a large number of previously reported transition metal systems.Future work in the area of main group catalysis in dehydrocoupling is likely to focus on the development of new types of E–E′ bond-forming reactions and the optimisation of catalyst activity. It can be noted in regard to the latter point that there have previously been no attempts to vary systematically the ligand set present in main group catalysts systems in order to optimise activity, as is the norm in the development of transition metal catalytic systems.1,14d As is already apparent from studies so far,4 changing the steric demands of the ligands in the precatalysts (Types A, B and C, Fig. 3) is one obvious way of modifying the kinetics and selectivity. However, a possible additional way of enhancing activity in the future is increasing the electronegativity of the ligands. The effects of this should be to increase the Lewis acidity of the metal centres, with the additional result of increasing redox stability; leading to greater polarisation of metal-bonded intermediates5 and potentially longer catalyst life-times.
Acknowledgements
We thank the EPSRC (DSW), The Leverhulme Trust (RJL, DSW), and Cambridge University (RLM).References
- J. P. Collman, L. S. Hegedus, J. R. Norton, R. C. Finke, Principles and Applications of Organotransition Metal Chemistry, University Science Books, U.S., 2nd revised edn 1987 Search PubMed.
- P. P. Power, Nature, 2010, 463, 171 CrossRef CAS.
- (a) P–H/P–H coupling; R. J. Less, R. L. Melen, V. Naseri and D. S. Wright, Chem. Commun., 2009, 4929 RSC; (b) V. Naseri, R. J. Less, M. McPartlin, R. E. Mulvey and D. S. Wright, Chem. Commun., 2010, 46, 5000 RSC; (c) M. S. Hill, M. F. Mahon and T. P. Robinson, Chem. Commun., 2010, 46, 2498 RSC.
- (a) Group 2 B–H/N–H coupling; J. Spielmann, G. Jansen, H. Bandmann and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 6290 CrossRef; (b) J. Spielman and S. Harder, J. Am. Chem. Soc., 2009, 131, 5064 CrossRef; (c) J. Spielmann, M. Bolte and S. Harder, Chem. Commun., 2009, 6934 RSC; (d) D. J. Liptrot, M. S. Hill, M. F. Mahon and D. J. MacDougall, Chem.–Eur. J., 2010, 16, 8508 CrossRef CAS; (e) J. Spielmann, D. F.-J. Piesik and S. Harder, Chem.–Eur. J., 2010, 16, 8307 CrossRef CAS; (f) M. S. Hill, M. Hodgson, D. J. Liprot and M. F. Mahon, Dalton Trans., 2011, 40, 7783 RSC; (g) P. Bellham, M. S. Hill, D. J. Liptrot, D. J. MacDougall and M. F. Mahon, Chem. Commun., 2011, 47, 9060 RSC.
- Group 3 B–H/N–H coupling; M. S. Hill, G. Kociok-Köhn and T. P. Robinson, Chem. Commun., 2010, 46, 7587 RSC.
- (a) Group 13 B–H/N–H coupling; H. J. Cowley, M. S. Holt, R. L. Melen, J. M. Rawson and D. S. Wright, Chem. Commun., 2011, 47, 2682 RSC; (b) M. M. Hansmann, R. L. Melen and D. S. Wright, Chem. Sci., 2011, 2, 1554 RSC.
- (a) Other catalytic reactions involving Group 2 metal reagents; M. R. Crimmins, M. Arrowsmith, A. G. M. Barrett, I. J. Casey, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2009, 131, 9670 CrossRef; (b) J. Spielmann and S. Harder, Eur. J. Inorg. Chem., 2008, 1480 CrossRef CAS; (c) J. Spielmann, F. Buch and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 9434 CrossRef CAS.
- (a) S. Greenberg and D. W. Stephan, Chem. Soc. Rev., 2008, 37, 1482 RSC; (b) D. W. Stephan, Angew. Chem., Int. Ed., 2000, 39, 314 CrossRef; (c) T. J. Clark, K. Lee and I. Manners, Chem.–Eur. J., 2006, 12, 8634 CrossRef CAS; (d) R. Waterman, Curr. Org. Chem., 2008, 12, 1322 CrossRef CAS; (e) R. Waterman, Dalton Trans., 2009, 18 Search PubMed.
- T. P. Fehlner, Inorganometallics, Plenum Press, New York 1992 Search PubMed.
- (a) C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279 RSC; (b) A. Staubitz, A. P. M. Robertson, M. E. Sloan and I. Manners, Chem. Rev., 2010, 110, 4023 CrossRef CAS.
- A further important issue in H2storage is regeneration of the unsaturated, dehydrogenated products (‘spent fuel’) back to amine boranes, see A. A. D. Sutton, A. K. Burrell, D. A. Dixon, E. B. Garner III, J. C. Gordon, T. Nakagawa, K. C. Ott, J. P. Robinson and M. Vasiliu, Science, 2011, 331, 1426 CrossRef CAS.
- V. Pons, R. T. Baker, N. K. Szymczak, D. J. Heldebrant, J. C. Linehan, M. H. Matus, D. J. Grant and D. A. Dixon, Chem. Commun., 2008, 6597 Search PubMed.
- L.-B. Han and T. D. Tilley, J. Am. Chem. Soc., 2006, 128, 13698 CrossRef CAS.
- (a) See T. M. Douglas, A. B. Chaplin, A. S. Weller and J. Am, J. Am. Chem. Soc., 2008, 130, 14432 CrossRef CAS; (b) R. Dalanegra, A. B. Chaplin and A. S. Weller, Angew. Chem., Int. Ed., 2009, 48, 6875 CrossRef; (c) A. B. Chaplin and A. S. Weller, Angew. Chem., 2010, 122, 591 CrossRef; (d) R. Dallanegra, A. P. M. Robertson, A. B. Chaplin, I. Manners and A. S. Weller, Chem. Commun., 2011, 47, 3763 RSC and references therein.
- R. Waterman, Organometallics, 2007, 26, 2492 CrossRef CAS.
- (a) M. C. Fermin and D. W. Stephan, J. Am. Chem. Soc., 1995, 117, 12645 CrossRef CAS; (b) N. Etkin, M. C. Fermin and D. W. Stephan, J. Am. Chem. Soc., 1997, 119, 2954 CrossRef CAS; (c) N. Etkin, M. C. Fermin and D. W. Stephan, Angew. Chem., Int. Ed., 2001, 40, 1865 CrossRef; (d) J. D. Masuda, A. J. Hoskins, T. W. Graham, C. Beddie, M. C. Fermin, N. Etkin and D. W. Stephan, Chem.–Eur. J., 2006, 12, 8696 CrossRef CAS.
- T. Beweries, S. Hansen, M. Kessler, M. Klahn and U. Rosenthal, Dalton Trans., 2011, 40, 7689 RSC.
- M. A. Beswick, N. Choi, C. N. Harmer, A. D. Hopkins, M. McPartlin and D. S. Wright, Science, 1998, 281, 1500 CrossRef CAS.
- M. McPartlin, R. L. Melen, V. Naseri and D. S. Wright, Chem.–Eur. J., 2010, 16, 8854 CrossRef CAS.
- J. L. Atwood, K. Robinson, F. R. Bennett, F. M. Elms, G. A. Koutsantonis and C. L. Raston, Inorg. Chem., 1992, 31, 2673 CrossRef CAS and references therein.
- (a) A. G. Avent, P. B. Hitchcock, A. V. Khvostov, M. F. Lappert and A. V. Protchenko, Chem. Commun., 2004, 2272 Search PubMed; (b) see also B. Luo, V. G. Young Jr. and W. L. Gladfelter, J. Organomet. Chem., 2002, 649, 268 CrossRef CAS.
- M. R. Crimmins, A. G. M. Bartlett, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2007, 26, 4076 CrossRef.
- J. Spielmann, D. Piesik, B. Wittkamp, G. Jansen and S. Harder, Chem. Commun., 2009, 45, 3455 RSC.
- S. Harder and J. Spielman, Chem. Commun., 2011, 47, 11945 RSC.
- (a) M. C. Denney, V. Pons, T. J. Hebden, D. M. Heinekey and K. I. Goldberg, J. Am. Chem. Soc., 2006, 128, 12048 CrossRef CAS; (b) T. J. Hebden, M. C. Denny, V. Pons, P. M. B. Piccoli, T. F. Koetzie, A. J. Schultz, W. Kaminsky, K. I. Goldberg and D. M. Heinekey, J. Am. Chem. Soc., 2008, 130, 10812 CrossRef CAS.
- C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424 CrossRef CAS.
- (a) A. Staubitz, A. P. Sato and I. Manners, Angew. Chem. Int. Ed., 2008, 47, 6212 CrossRef CAS; (b) A. Staubitz, M. E. Sloan, A. P. M. Robertson, A. Friedrich, S. Schneider, P. J. Gates, J. Schmedt auf der Günne and I. Manners, J. Am. Chem. Soc., 2010, 132, 13332 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |