Design of low-charge peptide sequences for high-yield formation of titania nanoparticles†
Chun-Xia
Zhao
*,
Lei
Yu
and
Anton P. J.
Middelberg
Centre for Biomolecular Engineering, Australian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, QLD, Australia. E-mail: z.chunxia@uq.edu.au; Fax: +61 7 3346 4197; Tel: +61 7 3346 4263
First published on 19th December 2011
Abstract
A series of low-charge peptides were designed to explore the functionality of different amino acids in precipitating precursor titanium(IV) bis(ammonium lactato)-dihydroxide (TiBALDH), and to obtain insight into the mechanism of TiO2 biomineralization.
The synthesis of inorganic nanomaterials using biomimetic approaches has attracted increasing interest owing to their benign synthesis conditions including neutral pH, ambient temperature, low pressure and the absence of caustic chemicals.1 As one of the major natural biomineralization materials, silica (SiO2) has received considerable attention in recent years. A wide range of organic templates have been utilized in the biomimetic mineralization of SiO2, including silicateins,2 silaffins,3 R5 peptide,4 phospholipids,5 collagen fibres,6 and peptide fibrils.7 By mimicking the process of biosilication, non-natural inorganic materials, such as iron oxide,8 noble metals,9 zinc oxide,10 germanium dioxide,11 and so on, have also been synthesized by harnessing various biotemplates. Among these materials, titanium dioxide (TiO2) is of special interest because of its attractive chemical, electrochemical, and optical properties and its widespread applications.
Various biomolecules have been exploited for the biomimetic synthesis of titania nanoparticles in solution at neutral pH and room temperature, including silicatein,12 R5 peptide,13 lysozyme,14 silaffins,15 polyamines,16 viruses,17 designed peptides,18 dendrimers11 and enzymes.19 The recombinant silaffin rSilC was used to induce the formation of rutile TiO2 under ambient conditions and neutral pH,15a whereas normally harsh conditions are required. Therefore, it is promising to develop new routes to synthesise titania-based materials under environmentally benign conditions by harnessing the biochemical mechanism of a biomimetic synthesis of TiO2 mediated by peptides or proteins. However, the molecular mechanisms underlying titania biomineralization are still unclear, and the understanding of how peptide or protein sequence and structure influence biomineralization is still not sufficient to realize the potential of biomimetic synthesis. In addition, current biomimetic synthesis strategies for TiO2 nanomaterials mainly focus on examining different biomolecules in a rather empirical way; little is known about the details of what kind of biotemplates are able to induce the formation of TiO2 having a desired crystal structure and morphology, and how these biomolecules regulate the nucleation and growth of TiO2 nanoparticles. Therefore, it becomes more crucial to identify the groups or active sites of proteins that facilitate the precipitation of titania, and to better understand the mechanisms involved in biomimetic synthesis.
Here, in an effort to obtain insight into the mechanism of TiO2 biomineralization and to better decouple the effects of precipitation activity and structure-directing function, we designed a series of peptides (Table 1), which are simpler than proteins in terms of sequence and structure, to explore the functionality of different amino acids in initiating the precipitation of precursor titanium(IV) bis(ammonium lactato)-dihydroxide (TiBALDH), and we use these to examine the biomineralization mechanism.
| Peptide | Sequence | PI | Charge at pH 7 | Specific activitya |
|---|---|---|---|---|
| a nmoles of TiO2 per nmole peptide after incubation for 20 min at room temperature. | ||||
| R5 | SSKKSGSYSGSKGSKRRIL | 11.22 | 5.9 | 6.45 ± 0.63 |
| Ti2 | RKKRKKRKKRKKGGGY | 12.04 | 11.9 | 29.58 ± 2.88 |
| Lac21E | Ac-MEELADS LEELARQ VEELESA CONH2 | 3.95 | −6.9 | n/a |
| Lac21K | Ac-MKKLADS LKKLARQ VKKLESA CONH2 | 9.74 | 5.1 | 34.58 ± 0.29 |
| Lac21 | Ac-MKQLADS LMQLARQ VSRLESA CONH2 | 8.03 | 1.1 | 1.70 ± 0.18 |
| AM1 | Ac-MKQLADS LHQLARQ VSRLEHA CONH2 | 8.08 | 1.6 | 27.11 ± 1.57 |
| AFD4 | Ac-MKQLADS LHQLAHQ VSHLEHA CONH2 | 7.02 | 0.1 | 1.48 ± 0.43 |
| P19 | Ac-PS ANSVAHS LANLAHS VSHLVSNAD CONH2 | 6.91 | −0.2 | n/a |
| P18 | Ac-PS ANSVARS LANLARS VSRLVSNAD CONH2 | 8.69 | 2.1 | 1.53 ± 0.90 |
| P17 | Ac-PS ANSVAKS LANLAKS VSKLVSNAD CONH2 | 8.59 | 2.1 | 6.94 ± 0.92 |
Two peptides, R513 (a biomimetic analogue derived from a repeat unit of the NatSil gene) and Ti2 (which derives from the TiO2 high-yielding peptide dTi-1(RKK)),18a were investigated first for their ability to precipitate TiO2 nanoparticles from the precursor TiBALDH. The control experiment showed that the TiBALDH precursor solution buffered to pH 7 by a sodium phosphate/citrate buffer was stable after 24 h incubation at room temperature in the absence of peptide. While both peptides (R5 and Ti2) were able to induce the formation of titania within seconds and in relatively high yield, peptide Ti2, with distinctly high arginine and lysine content, was superior. The precipitation activity of these two peptides was demonstrated to be consistent with previously published results.13,18a Following these benchmark experiments, two extreme peptides Lac21E20 and Lac21K20a were investigated. The glutamic acid-rich peptide Lac21E failed to induce the formation of any titania at neutral pH, while the precipitation activity was significantly enhanced by replacing the negatively charged glutamic acid with positively charged lysine (Lac21K), confirming literature results18a suggesting that the presence of positively-charged amino acids is essential for TiO2 precipitation. Three other peptides, Lac21, AM1 and AFD4, which have similar sequences, were then investigated. The parent sequence Lac21 is a well-studied synthetic peptide based on the tetramerization domain of the bacterial Lac repressor protein.21 AM1 differs from the Lac21 parent sequence through the substitution of two residues for histidine residues at positions 9 and 20. The AFD4 peptide includes two additional histidine residues replacing two arginine residues at positions 13 and 17.22 Lac21, having low positive charge at pH 7, showed only negligible precipitation activity, but with the incorporation of two histidine residues, AM1 exhibited surprisingly high titania precipitation activity, which was comparable to the lysine and arginine enriched Ti2 peptide, even though AM1 exhibits a small net positive charge (1.6) at pH 7. This result cannot be simplistically explained through previous findings that precipitation activity increases with the number of positive charges,18a as both Lac21 and AM1 have a similar pI. Interestingly, this result is not solely linked to histidine, as the addition of two further residues to AM1, giving sequence AFD4, causes an almost total loss of precipitation activity.
The last peptide group (P17, P18 and P19) was designed to further explore the independent roles of histidine, arginine and lysine in peptides having low positive charge relative to R5, Ti2 and Lac21K. Histidine-rich P19 did not induce the formation of any titania, even when incubated at room temperature for several days due to the net negative charge at pH 7. This is consistent with it having no net positive charge at this pH. Lysine-rich P17 and arginine-rich P18 showed very different precipitation activity even though they have the same number of positive charges. Lysine residues confer the strongest precipitation activity, followed by arginine and then histidine.
TiO2 precipitation assays with our designed peptides identified that the cationic amino acid residues (K, R and H) confer more activity for the biomimetic synthesis of TiO2 nanoparticles from TiBALDH at neutral pH, provided the peptides have high positive charge (e.g. Lac21K, R5 and Ti2). The presence of negatively charged amino acids (e.g. E) can completely inhibit the activity of TiO2-forming positively charged K, H and R. This finding confirms the hypothesis that cations are important to promote interaction with negatively charged TiBALDH precursors. Furthermore, the enrichment of lysine (K), arginine (R) and histidine (H) increased the titania precipitation activity, but the titania yield did not simply increase with the number of positive charges in the peptides, as has been suggested in the literature.18a
Specifically, the introduction of histidine residues into AM1 and AFD4 showed distinctive results in terms of the TiO2 precipitation activity, by firstly increasing the activity significantly when adding two histidines and then decreasing it dramatically with two more histidines. The results reveal a subtle and not understood role of histidine in TiO2 biomimetic synthesis. This new finding leads us to propose a specific TiBALDH catalysis mechanism at neutral pH, dependent on the functional hydroxy and imidazole side chains of serine and histidine which are present in AM1 (Scheme 1). Although it has been demonstrated that proteins or peptides can act as a general acid/base catalyst for the synthesis of silica from silicatein α,23 no peptide or protein has been previously designed to produce TiO2-based nanomaterials by utilizing this specific catalysis mechanism.
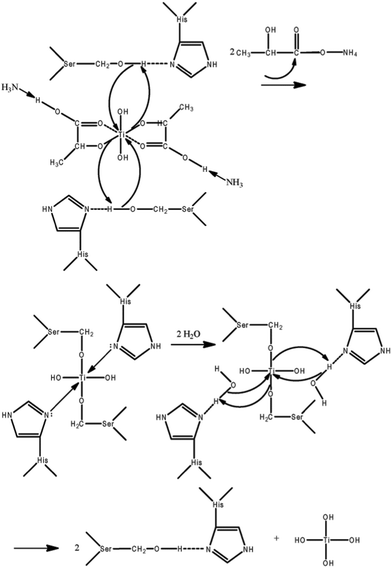 | ||
| Scheme 1 Serine-histidine catalysis for AM1-mediated hydrolysis of the TiBALDH precursor. | ||
In this study, as we propose above, AM1 was designed to contain two serine-histidine pairs. Briefly, AM1 molecules with positive charges bind to the negatively charged surface of TiBALDH due to simple electrostatic interaction. The hydroxy group in serine will attack the titanium atom in TiBALDH acting as a nucleophile, and a pair of electrons on the histidine nitrogen accepts the hydrogen from the serine hydroxyl group, thus coordinating the attack of the Ti atom. Nucleophilic attack on the titanium atom displaces ammonium lactate, and a transitory peptide–TiBALDH intermediate, which would be stabilized through a donor bond from the imidazole nitrogen atom, forms by releasing ammonium lactate. The addition of water hydrolyzes the Ti–O bond, and the hydroxy group in water attacks the titanium atom. Once again, the electrons on the histidine nitrogen accept the hydrogen from the hydroxyl group in water, and the bond between the oxygen of water and Ti is formed. Thus Ti(OH)4 is released and the serine-histidine pair is regenerated. Experimental results support this proposed mechanism of catalyzed synthesis of TiO2 nanoparticles, as AM1 showed distinctly high precipitation activity despite having a very low positive charge (1.6) at pH 7. AM1 activity is comparable to that of Ti2 which has positive charge of 11.9, confirming that the mechanism of AM1-templated TiO2 synthesis is different from that of Ti2.
Under the serine-lysine catalysis mechanism proposed here, AFD4, also having two serine-histidine pairs and two more histidine amino acids, would thus be expected to have a similar level of precipitation activity as AM1. However, AFD4 exhibited only very limited precipitation activity at pH 7. AFD4 carries 0.1 side-group positive charges at pH 7, which would inhibit the binding of AFD4 to the surface of TiBALDH, as this binding occurs only on surfaces which expose sufficient density of cationic groups, which tend to concentrate the anionic precursor creating local supersaturation. Therefore, positively charged peptides bind to the negatively charged TiBALDH molecules as a result of electrostatic attraction until the positively charged peptides accumulate to a certain limit, then the precursor TiBALDH becomes sufficiently close to be hydrolyzed by serine-histidine based catalysis. Therefore, we expect that under low net positive charge (like 0.1 for AFD4), the electrostatic interaction is not sufficient to bring the precursor and peptide molecules close enough to initiate synthesis. Based on this hypothesis, we would expect that the precipitation activity of AFD4 will be restored at pH 6.45, where AFD4 has the same positive charge (1.6) as AM1 does at pH 7. This predicted result was confirmed experimentally, and the precipitation activity of AFD4 at pH 6.45 is 26.28 ± 2.4, which nearly equals that of AM1 (27.11 ± 1.57). Therefore, the specific serine-histidine catalysis mechanism has been further confirmed with direct experimental evidence, while also highlighting the need for sufficient positive charge to attract the anionic TiBALDH precursor. This finding provides further support for the serine-histidine catalysis mechanism proposed here.
The TiO2 precipitates obtained by these designed peptides were examined by transmission electron microscopy (TEM) (Fig. 1), high-resolution TEM (HR-TEM), selected-area electron diffraction (SAED) and energy dispersive X-ray spectroscopy (EDXS) (ESI†). None of these peptides are able to control the shape and crystal structure of TiO2 nanoparticles due to the lack of structural complexity and polyfunctionality found in proteins, therefore, they are unable to reproduce the shape-controlling activity of silaffin-producing rutile TiO2.15a For example, even though AM1 can rapidly precipitate TiO2 under mild conditions, it is not able to promote the growth of TiO2 nanoparticles under the conditions reported here, and thus the particle morphology, due to the lack of specific binding of AM1 to TiO2 nanoparticles. The zeta potential of TiO2 nanoparticles was measured during the precipitation process. After precipitation, a zeta potential of −35.5 mV was recorded by directly measuring the precipitation solution. After washing the nanoparticles with Milli-Q water, the zeta potential decreased from −35.5 to −10.9 mV due to the removal of unreacted TiBALDH. Afterwards, the TiO2 nanoparticles were mixed with 200 μM AM1 solution, and the zeta potential increased to 17.4 mV, while it dropped to −9.89 mV after washing with Milli-Q water twice. Furthermore, the EDXS results (Fig. S2†) indicated that AM1-induced precipitates contain titanium, oxygen, and phosphorus. The absence of sulfur indicates that AM1 was not incorporated or bound extensively to TiO2 nanoparticles, as AM1 contained one residue of the sulfur-bearing amino acid, methionine (M). Therefore, the possibility that AM1 had already saturated nanoparticle binding sites and remained bound through the washing steps can be ruled out. Therefore, it was confirmed that AM1 was not able to tightly bind to TiO2 nanoparticles in a specific fashion. As a result, AM1 appears unable to regulate nanoparticle formation through action as a stabilizer or capping agent.
 | ||
| Fig. 1 TEM images of titania formation by different peptides. Scale bars are 200 nm. | ||
Conclusions
A deeper understanding of the biomineralization mechanism of peptide-enabled titania nanoparticle synthesis using the precursor TiBALDH was obtained by investigating the precipitation activity of various peptides with different amino acid sequences. A specific serine-histidine catalysis mechanism was experimentally suggested. Firstly, electrostatic interaction brings the peptide closer and promotes binding of the precursor TiBALDH, which supports the hypothesis that polycations are important for interacting with negatively charged precursors. Then, the functional hydroxy and imidazole side groups of serine and histidine facilitate the specific catalysis of TiBALDH at neutral pH. Based on this mechanism, peptides AM1 and AFD4, with low charge and at pH 7 and pH 6.4, respectively, are able to produce titania nanoparticles at high precipitation activity. Better understanding of the relationship between the peptide sequence and biomineralization ability, the molecules and mechanisms of biomimetic synthesis, will enable further design of novel peptides with selected properties of interest in functional materials synthesis, and will provide new approaches for producing titania-based functional materials.Acknowledgements
This work was funded by Australian Research Council grant DP1093056. Dr Chun-Xia Zhao acknowledges support from the Australian Research Council in the form of an Australian Postdoctoral Fellowship (DP110100394).References
- (a) C. L. Chen and N. L. Rosi, Angew. Chem., Int. Ed., 2010, 49, 1924 CAS; (b) M. B. Dickerson, K. H. Sandhage and R. R. Naik, Chem. Rev., 2008, 108, 4935 CrossRef CAS.
- (a) J. N. Cha, K. Shimizu, Y. Zhou, S. C. Christiansen, B. F. Chmelka, G. D. Stucky and D. E. Morse, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 361 CrossRef CAS; (b) K. Shimizu, J. Cha, G. D. Stucky and D. E. Morse, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6234 CrossRef CAS.
- N. Kroger, R. Deutzmann and M. Sumper, Science, 1999, 286, 1129 CrossRef CAS.
- M. R. Knecht and D. W. Wright, Chem. Commun., 2003, 3038 RSC.
- S. Baral and P. Schoen, Chem. Mater., 1993, 5, 145 CrossRef.
- Y. Ono, K. Nakashima, M. Sano, J. Hojo and S. Shinkai, Chem. Lett., 1999, 1119 CrossRef CAS.
- (a) V. M. Yuwono and J. D. Hartgerink, Langmuir, 2007, 23, 5033 CrossRef CAS; (b) J. E. Meegan, A. Aggeli, N. Boden, R. Brydson, A. P. Brown, L. Carrick, A. R. Brough, A. Hussain and R. J. Ansell, Adv. Funct. Mater., 2004, 14, 31 CrossRef CAS.
- (a) M. Allen, D. Willits, J. Mosolf, M. Young and T. Douglas, Adv. Mater., 2002, 14, 1562 CrossRef CAS; (b) A. Sinha, S. Nayar, G. V. S. Murthy, P. A. Joy, V. Rao and P. Ramachandrarao, J. Mater. Res., 2003, 18, 1309 Search PubMed.
- J. M. Slocik and D. W. Wright, Biomacromolecules, 2003, 4, 1135 CrossRef CAS.
- M. Umetsu, M. Mizuta, K. Tsumoto, S. Ohara, S. Takami, H. Watanabe, I. Kumagai and T. Adschiri, Adv. Mater., 2005, 17, 2571 CrossRef CAS.
- S. L. Sewell, R. D. Rutledge and D. W. Wright, Dalton Trans., 2008, 3857 RSC.
- J. L. Sumerel, W. J. Yang, D. Kisailus, J. C. Weaver, J. H. Choi and D. E. Morse, Chem. Mater., 2003, 15, 4804 CrossRef CAS.
- S. L. Sewell and D. W. Wright, Chem. Mater., 2006, 18, 3108 CrossRef CAS.
- H. R. Luckarift, M. B. Dickerson, K. H. Sandhage and J. C. Spain, Small, 2006, 2, 640 CrossRef CAS.
- (a) N. Kroger, M. B. Dickerson, G. Ahmad, Y. Cai, M. S. Haluska, K. H. Sandhage, N. Poulsen and V. C. Sheppard, Angew. Chem., Int. Ed., 2006, 45, 7239 CrossRef CAS; (b) E. Kharlampieva, J. M. Slocik, S. Singamaneni, N. Poulsen, N. Kroger, R. R. Naik and V. V. Tsukruk, Adv. Funct. Mater., 2009, 19, 2303 CrossRef CAS.
- K. E. Cole and A. M. Valentine, Biomacromolecules, 2007, 8, 1641 CrossRef CAS.
- M. T. Klem, M. Young and T. Douglas, J. Mater. Chem., 2008, 18, 3821 RSC.
- (a) M. B. Dickerson, S. E. Jones, Y. Cai, G. Ahmad, R. R. Naik, N. Kroger and K. H. Sandhage, Chem. Mater., 2008, 20, 1578 CrossRef CAS; (b) Y. Fang, N. Poulsen, M. B. Dickerson, Y. Cai, S. E. Jones, R. R. Naik, N. Kroger and K. H. Sandhage, J. Mater. Chem., 2008, 18, 3871 RSC.
- G. Chen, M. Li, F. Li, S. R. Sun and D. G. Xia, Adv. Mater., 2010, 22, 1258 CrossRef CAS.
- (a) R. Fairman, H. G. Chao, T. B. Lavoie, J. J. Villafranca, G. R. Matsueda and J. Novotny, Biochemistry, 1996, 35, 2824 CrossRef CAS; (b) A. P. J. Middelberg, L. He, A. F. Dexter, H. H. Shen, S. A. Holt and R. K. Thomas, J. R. Soc. Interface, 2008, 5, 47 CrossRef CAS.
- R. Fairman, H. G. Chao, L. Mueller, T. B. Lavoie, L. Y. Shen, J. Novotny and G. R. Matsueda, Protein Sci., 1995, 4, 1457 CAS.
- A. F. Dexter and A. P. J. Middelberg, J. Phys. Chem. C, 2007, 111, 10484 CrossRef CAS.
- Y. Zhou, K. Shimizu, J. N. Cha, G. D. Stucky and D. E. Morse, Angew. Chem., Int. Ed., 1999, 38, 779 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures. See DOI: 10.1039/c2ra00726f |
| This journal is © The Royal Society of Chemistry 2012 |