ZnO nanoparticle-coated surfaces inhibit bacterial biofilm formation and increase antibiotic susceptibility†
Guy
Applerot
a,
Jonathan
Lellouche
ab,
Nina
Perkas
a,
Yeshayahu
Nitzan
b,
Aharon
Gedanken
*a and
Ehud
Banin
*b
aDepartment of Chemistry and Kanbar Laboratory for Nanomaterials, Center for Advanced Materials and Nanotechnology, Bar-Ilan University, Ramat-Gan 52900, Israel. E-mail: gedanken@mail.biu.ac.il; Fax: +972-3-7369928; Tel: +972-3-5318315
bThe Biofilm Research Laboratory, Center for Advanced Materials and Nanotechnology, The Mina and Everard Goodman Faculty of Life Sciences, Bar-Ilan University, Ramat-Gan 52900, Israel. E-mail: ehud.banin@biu.ac.il; Fax: +972-3-7384053; Tel: +972-3-5317288
First published on 26th January 2012
Abstract
Nanotechnology is providing new ways to manipulate the structure and chemistry of surfaces to inhibit bacterial colonization. In this study, we evaluated the ability of glass slides coated with zinc oxide (ZnO) nanoparticles to restrict the biofilm formation of common bacterial pathogens. The generation of hydroxyl radicals, originating from the coated surface, was found to play a key role in antibiofilm activity. Furthermore, we evaluated the ability of the nanoparticle coating to enhance the antibacterial activity of commonly-used antibiotics. The ZnO nanoparticles were synthesized and deposited on the surface of glass slides using a one-step ultrasound irradiation process. Several physico-chemical surface characterization methods were performed to prove the long-term stability and homogenity of the coated films. Collectively, our findings may open a new door for utilizing ZnO nanoparticle films as antibiofilm coating of surfaces, thus providing a versatile platform for a wide range of applications both in medical and industrial settings, all of which are prone to bacterial colonization.
Introduction
Bacterial attachment to surfaces leading to the formation of communities of bacterial cells is a major problem in many diverse settings. This sessile community of microorganisms, also termed a biofilm, is attached to an interface, or to each other, and embedded in an exopolymeric matrix. It manifests an altered growth rate and transcribes genes that free-living microorganisms do not transcribe.1 Probably the most characteristic phenotype of the biofilm mode of growth is its inherent resistance to antimicrobial treatment and immune response killing.2 Medical implants and in-dwelling devices are especially prone to bacterial colonization and biofilm formation. Thus, conventional antibiotic therapy against device-associated biofilm organisms often fails without the removal of the infected implant.2–4 In fact, it has been estimated that the number of implant-associated infections approaches 1 million/year in the US alone, and their direct medical costs exceed $3 billion annually.3–6 The inherent resistance of biofilms to killing and their pervasive involvement in implant-related infections has prompted research in the area of biocidal surfaces/coatings.7,8 Such antibiofilm coatings may also be in use for various industrial applications such as drinking water distribution systems and food packaging. Hence, searching for the next generation of materials which can inhibit bacterial colonization and may serve as disinfecting agents is called for.9 In this respect, insoluble ceramic powders of metal oxides such as ZnO or MgO have been found to exhibit marked antibacterial activity.10–12 Utilizing ZnO as an inorganic antibacterial agent has a major advantage due to its ability to withstand harsh processing, compared to organic materials such as conventional antibiotics.13ZnO is also widely used as an active ingredient for dermatological applications in creams, lotions and ointments on account of its antibacterial properties.14,15 Moreover, research in the area of nanometre-scaled metal oxides in general, and ZnO in particular, has demonstrated a clear size dependence of various properties, e.g., electromagnetic, optical, catalytic, as well as antibacterial activities.10,11,16–19 The rapid development of different methods for the fabrication and deposition of nanomaterials on polymer and glass surfaces significantly enhanced their application in electronic devices and biotechnology. Recently, some low temperature methods for the deposition of nanoparticles (NPs) on a glass substrate were reported, e.g., electrode plating of spin-coated NPs and the deposition of NPs on modified glass slides.20,21 However, as the utilization of chemicals leads to environmental concerns, other approaches that concur with simplicity and are environmentally friendliness are desired.Sonochemical irradiation has been proven as an effective technique for the synthesis of nanophased materials, as well as for their deposition on various polymeric matrices in a one-step process.22 Furthermore, sonochemical irradiation fulfils the requirement of minimizing the use of toxic chemicals, solvents, energy, etc. and therefore is regarded as a “green” chemistry approach. The chemical effects of ultrasound arise from acoustic cavitation phenomena, i.e., the formation, growth, and implosive collapse of bubbles in a liquid medium. In this technique, when the created bubbles collapse near a solid surface, they produce enormous amounts of energy from the conversion of the kinetic energy of the motion of the liquid into heating the contents of the bubble. The compression of the bubbles during cavitation is more rapid than the thermal transport, which generates short-lived, localized hotspot bubbles with a temperature of around 5000 K, a pressure of roughly 1000 atm, and heating and cooling rates above 1 × 1010 K s−1. High-velocity fluid agitation and shock waves are also created during the compression of the bubbles near solid surfaces. These energetic jets throw the newly-synthesized NPs onto the surface at a very high speed (>100 m s−1), causing the NPs to adhere strongly to the solid surface. 23,24
Previous works demonstrated the use of sonochemistry as a perspective method to coat various substrates such as paper,25 glass surfaces,26 and fabrics27 with ZnO NPs. The activity of ZnO NP-coated surfaces as an antibacterial agent was investigated and their excellent bactericidal effect was demonstrated on the free-living bacterial community. In the present work, we took several steps forward and evaluated the antibiofilm activity of the coated surfaces. The antibiofilm tests were performed using two common nosocomial biofilm-forming pathogens, i.e., Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus). In addition, in the present study, using electron spin resonance (ESR) coupled with the spin-trapping technique and attenuated total reflectance-Fourier transform infrared (ATR-FTIR), we demonstrate the formation of reactive oxygen species (ROS) emerging from the coated glass surface. We also were able to show that a short pre-exposure to the ZnO NP-coated surface enhances the susceptibility of bacteria to antibiotic activity. Finally, supplementary physicochemical and surface characterization methods such as focused ion beam (FIB) assisted cross-sectional analysis, time-of-flight secondary ion mass spectrometry (ToF-SIMS), and Rutherford backscattering spectrometry (RBS), were added as a continuance to our previous work to verify the long-term stability of the coated film.
Experimental
Coating procedure.26 All the reagents were purchased from Sigma Aldrich and were of analytical chemical purity and used without additional purification. The ZnO NPs were deposited on a glass slide by the sonochemical irradiation of the Zn2+ ions' precursor in a water–ethanol solution. This solvent is considered an environmentally-friendly solution. Zinc acetate tetrahydrate, Zn(Ac)2·4H2O, was used as a precursor for this reaction. Typically, the zinc acetate was dissolved in a water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol = 1:9 mixture to obtain a 0.05 mol L−1 concentration of Zn2+ ions. The pH of the solution was adjusted with the addition of ammonia to pH 8. The glass slide was then inserted into the reactor and the sonication was done with an immersed Ti-horn (20 kHz, 750W at 70% efficiency) for a fixed period of time. After the reaction the sonochemically-coated glass was washed with water and ethanol and allowed to dry in air at room temperature. The ZnO content on the glass was determined by volumetric titration with ethylenediaminetetraacetic acid (EDTA) after treatment of the sample in a 0.5 M solution of HNO3.
:ethanol = 1:9 mixture to obtain a 0.05 mol L−1 concentration of Zn2+ ions. The pH of the solution was adjusted with the addition of ammonia to pH 8. The glass slide was then inserted into the reactor and the sonication was done with an immersed Ti-horn (20 kHz, 750W at 70% efficiency) for a fixed period of time. After the reaction the sonochemically-coated glass was washed with water and ethanol and allowed to dry in air at room temperature. The ZnO content on the glass was determined by volumetric titration with ethylenediaminetetraacetic acid (EDTA) after treatment of the sample in a 0.5 M solution of HNO3.
Measurements of the coating thickness and nanoparticle penetration depth into the bulk of the glass were done by a focused ion beam (FIB) using a FEI Helios 600 system.
ToF-SIMS analysis was conducted by a time-of-flight secondary ion mass spectrometer (PHI TRIFT II) on ZnO-coated glass slides. The Ga+ primary ion beam was operated at 25 keV and 20 nA. Positive secondary ion spectra were acquired from 50 × 50 μm2 areas of the surfaces. Sputtering was performed by Ga+ ions for 1 s followed by spectrum acquisition. The sputtering area is 150 × 150 μm2.
Rutherford Backscattering Spectroscopy (RBS) microbeam analysis was performed with a 3.0 MeV He+ beam generated by a Tandetron 1.7 MV accelerator of High Voltage Engineering Europe. The conditions of RBS analysis on the microbeam scanning system (model OM 2000, Oxford Micro beams, Ltd.) were: spatial resolution, about 2 μm; current density, 1 nA; mapping area of 500 × 500 μm2. The analysis of results was done by converting the units of at/cm2 to nm using the weighted mean of the atomic densities of detected zinc oxide at the surface of the sample, which “diffused” into the glass film.
Spin trapping measurements, coupled with electron spin resonance (ESR) spectroscopy detection, used the ESR spin-trapping technique utilizing the spin trap 5,5-dimethyl-1-pyrroline-N-oxide (DMPO, 0.02 M) (Sigma, St. Louis, MO). The aqueous medium in which square pieces (1 cm × 1 cm) of a ZnO-coated glass slide were placed and the appropriate spin trap were drawn by a syringe into a gas permeable Teflon capillary (Zeus Industries, Raritan, NJ) and inserted into a narrow quartz tube that was kept open at both ends. The tube was then placed in the ESR cavity and the spectra were recorded on a Bruker ESR 100d X–band spectrometer. The ESR measurement conditions were as follows: frequency, 9.74 GHz; microwave power, 20 mW; scan width, 65 G; resolution, 1024; receiver gain, 2 × 105; conversion time, 82 ms; time constant, 655 ms; sweep time, 84 s; scans, 2; modulation frequency, 100 kHz. After acquisition, the spectrum was processed using Bruker WIN-ESR software version 2.11 for baseline correction. The peak intensity was calculated by double integration of the peak signals, and the intensity was expressed in arbitrary units.
Attenuated total reflectance-Fourier transform infrared (ATR-FTIR) was obtained on a Bruker Vector 22 spectrometer. Spectra of the as-deposited films were collected using a 60 × 20 × 0.45 mm Si parallelogram prism prepared in-house by polishing the two short edges of a freshly cut, double-side polished silicon wafer to a 45° angle. The background data were collected following piranha treatment of the cleaned ATR prism, and the sample data were collected after the deposition of the monolayer. The background spectrum of the clean ATR prism was subtracted from each sample spectra. Typically, 44 sample scans were collected, at a nominal resolution of 4 cm−1.
For the microscopic examination of biofilm formation, a flow cell system was implemented. The biofilm system is composed of a polycarbonate chamber into which the tested glass coupons (ZnO coated and uncoated) are inserted. Using a Watson Marlow peristaltic pump and silicon manifold tubing (0.8 mm diameter), the growth medium is pumped at a constant rate (10 ml h−1) through the chamber. The flow cell was initially inoculated with a 0.3 OD595cell culture of either E. coli or S. aureus (this approximately corresponds to 3×108CFU ml−1 (CFU; colony-forming units)) under domestic lamplight. The flow was initiated after 1 h incubation at room temperature (flow rate of 10 ml h−1), and 1% TSB or 1% TSB-Glu (diluted in double distilled water; DDW) was used as a growth medium for E. coli and S. aureus, respectively. After 24 h the glass slides were removed from the experimental flow cell and washed with DDW to remove unattached cells. For imaging, the slides were stained using the Live/Dead BacLight kit (Molecular Probes, Invitrogen, manufacturer protocol). A SYTO9/propidium iodide mixture stain was dissolved in a mixture of 3% DMSO (dimethylsulfoxide) and DDW (15 min incubation). Viable bacteria with intact cell membranes are stained in green, whereas dead bacteria with damaged membranes are stained in red. Both excitation/emission maxima for these two dyes are 480/500 nm for the SYTO9 stain, and 490/635 nm for the propidium iodide. Biofilm formation was monitored using a confocal scanning laser microscope (Leica SPE, San Diego, California, United States). Obtained images were further processed by the Imaris Image Analysis software (Imaris v.6.0, Bitplane Scientific Software) and represent the general trend seen in three independent experiments. The biofilm biomass was also quantified by a viable count. Glass slides washed with DDW and the biofilm cells were detached by exposure to a low energy sonication water bath (TRANSSONIC 460, ELMA) for 1 min and centrifuged at 4000 rpm for 5 min to form pellet cells. Cells were re-suspended and serial dilutions were plated on Luria Bertani (Difco) agar plates to enumerate the viable cells. The experiment was conducted in triplicate and was repeated three times independently. The results were found to be reproducible (P < 0.05).
Minimal inhibitory assay. Test tubes containing a nutrient broth with different concentrations of purified antibiotics were inoculated with 100 μl of the bacterial suspension (105CFU ml−1). Inoculated tubes were incubated for 24 h at 37 °C and growth was observed by measuring OD at 660 nm. MIC is the minimal antibiotic concentration at which no growth was detected. All tests were carried out in duplicate.
The antibiotics used in this study included chloramphenicol (CP), nalidixic acid (NA), Ampicillin (AM), for both stains; Erythromycin (ER) for E. coli 1313 and Tobramycin (TO) for S. aureus 195 (Both bacterial isolates were obtained from the Bacteriological Laboratory of the Meir Hospital, Kfar Sava, Israel28). Two types of agar medium were prepared: (1) an agar medium without antibiotics that was used as a “non-selective medium”; (2) an agar medium containing one of the antibiotics at half of the MIC, termed “selective medium”. A typical procedure was as follows: cultures of the bacteria were grown on nutrient agar (Difco, Detroit, MI) overnight. These cultures were then transferred into a nutrient broth (NB) at an initial optical density (OD) of 0.1 at 660 nm and allowed to grow at 37 °C with aeration. When the cultures reached an optical density of 0.3 OD at 660 nm (the onset of the logarithmic phase), they were centrifuged and washed twice with saline at pH 6.5 to yield a final bacterial concentration of approximately 108CFU ml−1. An aliquot of 4.5 ml of a saline solution containing the impregnated glass (sized 1 × 1 cm) was poured into a vial with an inner diameter of 2.5 cm. 500 μl of the strain cells was then pipetted into the vial. The initial bacterial concentration in the vial was approximately 107CFU ml−1. Bacterial suspensions were incubated within the saline solution containing the impregnated glass and shaken at 150 rotations a minute at 37 °C for up to 10 min. Samples, each of 100 μl, were taken at a specified time, diluted tenfold in saline, and plated onto the selective and non-selective nutrient agar plates. The plates were allowed to grow for 24 h at 37 °C, and then counted for viable bacteria. The viable bacteria were monitored by counting the number of CFU from the appropriate dilution on nutrient agar plates, and comparing the number of colonies on the selective and non-selective media. To ensure that any decrease in bacterial number was likely to be due to exposure to coated-glass treatment, two controls were included in the experiment: the first with saline (without any glass), and the second with saline with an uncoated glass. In these tests, the plating out of each dilution was duplicated, and the whole assay was repeated five times independently. The results were found to be reproducible (P < 0.05).
Results and discussion
Coating mechanism
The coating process involves the in situ generation of ZnO NPs and their subsequent deposition on glass slides in a one-step reaction via ultrasound irradiation. ZnO NPs were formed during irradiation according to the sonohydrolysis mechanism, as discussed previously.26,29 In our case, microjets and shock waves, which are known to be created after the collapse of the acoustic bubbles near surface solids, promote the fast migration of the newly-formed ZnO NPs to the glass surface. In this regard, a physical embedment of the NPs on the substrate may be the reason underlying the particles' strong adherence to the glass. A concise illustration of the mechanism for the sonochemical deposition of NPs is schematically presented in Fig. 1. It should also be emphasized that the coating was stable and could not be removed by a simple washing procedure with water, ethanol, and/or acetone. The methods we used for the leaching examination were DLS and transmission electron microscopy (TEM) after placing the coated glasses in each of the above-mentioned solvents for a duration of over 7 days. The DLS and TEM studies did not reveal the presence of any NPs in the leaching solution.26 That means that the sonochemically-deposited ZnO NPs are strongly anchored to the glass substrate.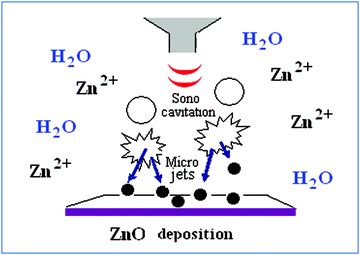 | ||
| Fig. 1 Scheme of the sonochemical deposition of ZnO NPs on the solid substrate. | ||
Morphology characterization
In our previous work, the deposited glass film was analyzed using characterization methods such as XRD, SEM, AFM, and optical spectroscopy.26SEM images demonstrated the morphology of the coated layer on the glass. The ZnO NPs deposited on the glass were a spherical shape resulting in a 0.65% (wt.) ZnO coating. The surface morphology of the deposited ZnO was also studied by AFM, revealing a multilayer growth of the ZnO coating. In this study, we determined the coating thickness using FIB-assisted, cross-sectional analysis. During the FIB operation, concentrated positive gallium ions (Ga+) are accelerated into an insulating material and they etch off any exposed surface. The signal from the sputtered ions or secondary electrons is collected to form an image.Since the sample preparation procedure requires the deposition of platinum over the sample prior to sectioning by FIB, the measured samples show a rather smooth metallic layer over the top of the deposited platinum. At the edges of the sections, the ZnO can be seen as the bright regions beneath the platinum layer. Notably, FIB analysis of the sample (Fig. 2) showed the presence of more than one layer of ZnO coating under the platinum layer, with a thickness of 40–70 nm for each layer. The NPs were deposited very close to each other, forming a continuous layer. This was also in accordance with the SEM and AFM results, which verified a relative homogenous deposition.
 | ||
| Fig. 2 FIB cross-sectional images of ZnO-coated glass (that was covered for protection by platinum). a) The scale bar is 300 nm. b) A closer examination of the ZnO layers sized 40–70 nm; the scale bar is 400 nm. | ||
Depth profile
As we pointed out above, the adhesion nature of the sonochemically-deposited zinc oxide NPs to the glass was steady in the coated samples and could not be removed by washing. Therefore, the question arose as to whether we achieved only surface deposition, or whether the zinc oxide NPs also penetrated the glass. Hence, to characterize the penetration profile of ZnO NPs onto a glass surface, ToF-SIMS and RBS were employed. The analysis was performed on ZnO-coated glass and the result of the surface mass spectrum ToF-SIMS measurement is shown in Figure S1 ESI.† First, the distribution of the ZnO NPs on the glass surface was further confirmed by a surface mass spectrum of the coated sample, (Figure S1a). As expected,25 ion peaks corresponding to 64Zn+, 64ZnH+, 66Zn+, 66ZnH+, 68Zn+, and 68ZnH+, were observed. Concerning the penetration depth, as can be seen in Figure S1b, ESI,† at the depth of the first few nanometres, a very sharp increase in the glass components (Si, Al, and Mg) is detected, while their quantity remains unchanged along the width of the glass. In contrast, the amount of zinc is much larger at the first few nanometres than the amount of the glass components. At a depth of 10–60 nm, the amount of zinc is reduced until it reaches the detection limit at approximately 80 nm. This result indicates that a major amount of zinc oxide is embedded within the glass. Another depth profile analysis was performed by RBS. RBS is an analytical nuclear technique to determine depth profiles of elemental concentration. The RBS technique is based on elastic collisions between ions and the atomic nucleus.30 The slowing down of ions in matter provides a depth resolution that is beyond the detection limit of ToF SIMS. The results show that small amounts of ZnO NPs were inserted into the glass in a depth of even more than 200 nm (Fig. 3). The stoichiometry of glass components seems to be slightly modified when the ZnO concentration is higher and represents up to 20% of the composition (at the surface). However, at the deep layers, the glass film remains unchanged.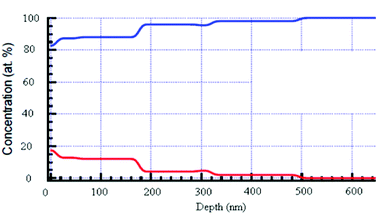 | ||
| Fig. 3 RBS of a glass slide coated with ZnO NPs. The red line is the ZnO NPs and the blue line stands for the glass components (Si, O, Na, Mg). No signal of ZnO was detected for the uncoated glass (data not shown). | ||
ROS detection
To characterize ROS formation by ZnO-glass coated films, we used the ESR spin-trapping technique coupled with a DMPO spin trap. A piece of the coated glass was deposited in water in the presence of the spin trap and introduced into the ESR cavity, as described in the experimental section, and the ESR spectrum was recorded. A typical spectrum of a characteristic DMPO–OH spin adduct with four resolved peaks was detected, as depicted in Fig. 4, and was in good agreement with our previous results obtained for ROS produced in a water suspension containing ZnO NPs.11,31 Following the ESR results, it is evident that hydroxyl radical species, originating from water molecules adsorbed on the defect sites surface of the ZnO NP coating, play a critical role in generating the antibacterial action of the coated glass. However, it should be kept in mind that our bacteriological assays and the ESR detection were all performed in an aqueous environment. Utilizing ZnO NP coating for dryer applications calls for an examination as to whether conditions of exposure to the ambient atmosphere are enough for adsorbing enough water molecules at the surface of the metal oxide to generate •OH.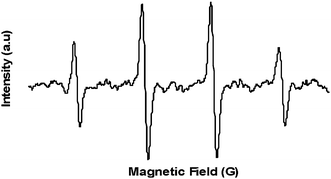 | ||
| Fig. 4 Spectrum of DMPO–OH derived from ZnO NP coating in the presence of the spin trap, DMPO. | ||
To address this, we performed attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy at ambient air settings, looking for on-surface absorbed hydroxyl groups. Interestingly, the characterization depicted in Figure S2 ESI,† showed a band at ∼1600 cm−1 that could be attributed to the absorbed water originating from ambient moisture, on the substrate surface. A broad band (3100–3700 cm−1) centered at around 3400 cm−1, characteristic of a –OH functional group (free and hydrogen bonded), was also observed. These peaks did not exist for the uncoated substrate.24,32,33
Biofilm inhibition
The effectiveness of ZnO NP-coated surfaces to inhibit bacterial colonization as a function of time was examined. Using a continuous flow chamber, the glass slides were challenged with bacterial cultures for ten consecutive days. Several coupons were removed each day to quantify the biofilm that had developed thus far. This protocol allowed us to unambiguously test the ability of the ZnO coating to generate sufficient antibiofilm activity over time. A representative selection of images obtained from the confocal laser-scanning microscope (CLSM) is presented in Fig. 5.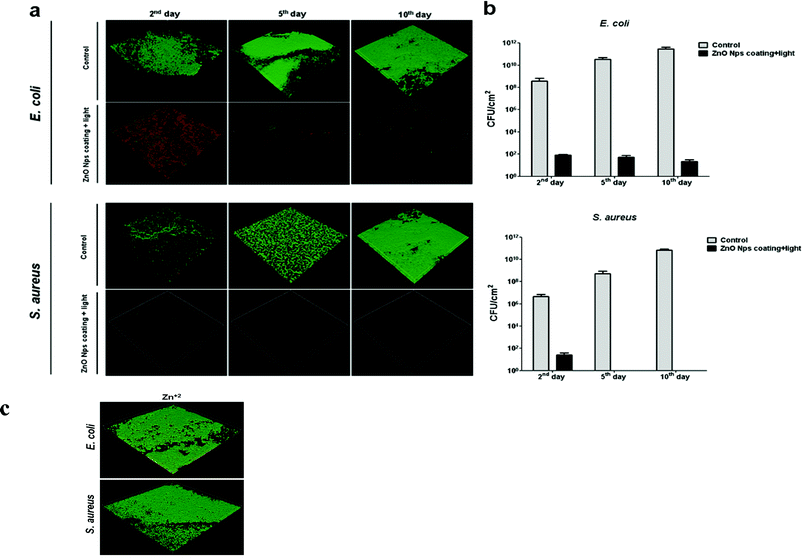 | ||
| Fig. 5 Antibiofilm activity of ZnO NP coating on glass surfaces. a) CLSM images showing results from bacteria (E. coli and S. aureus) viability stains; the green and red stains respectively indicate living bacteria and membrane-compromised bacteria. Images obtained by confocal laser scanning microscopy represent the general trend seen in three independent experiments. b) Viable count of the biofilm cells. Control refers to the biofilm development on uncoated surfaces. Data represent the mean values of three independent experiments conducted in triplicate. c) Effect of Zn+2 on biofilm development. E. coli and S. aureus grown for 10 days in flow cells on glass surfaces (experimental setup and growth conditions described in the Methods section) with Zn+2 (50 mg L−1). Green and red staining represents live and dead bacterial cells, respectively. | ||
The results of this viability test imply that ZnO glass coating inhibits biofilm for both E. coli and S. aureus, consistent with previous reports of its antibacterial properties.
Generally, sparsely-distributed microbes, many of which are dead (the red dots in Fig. 5a) appear on the coated glass slides, compared with the unmodified glass slides which allowed the formation of a continuous biofilm layer (stained green; control). As depicted in Fig. 5b, these data are also supported by viable counts obtained directly from the biofilm formed on the surfaces. Uncoated glass surfaces supported a massive biofilm formation (∼3.2 × 1011 and ∼5.8 × 1010CFU cm−2 for E. coli and S. aureus, respectively, for the 10th day), while ZnO-coated surfaces dramatically restricted bacterial colonization (∼20 and ∼0 CFU cm−2 for E. coli and S. aureus respectively, on the last day). It is noteworthy that we have already excluded the possibility that zinc ions existing in the ZnO NPs aqueous suspension were responsible for the antibacterial activity11 (see also ref. 12). In the current study, we conducted a control experiment with Zn2+ at a concentration of 50 mg L−1, which is more than 10 times the solubility of ZnO. No effect on the biofilm growth could be observed for either bacterial strain (∼1.8 × 1010 and ∼3.4 × 1010CFU cm−2 for E. coli and S. aureus, respectively, for the 10th day; Fig. 5c). We also examined the viability of planktonic bacteria in the chamber to exclude killing during the incubation time before flow activating. No significant reduction could be observed when ∼1.2 × 106CFU ml−1 and ∼2.3 × 106CFU ml−1 were counted for E. coli and S. aureus, respectively. Hence, the reduction in biofilm formation was likely related to the reduction in bacterial proliferation on the coated surface caused by exposure to the NPs rather than to dissolved ions. Taking into consideration the continuous bacterial challenge of the coated glass slides during the experiment, these results further confirm the long-term stability and effectiveness of the coating as it does not lose its antibacterial effect over time due to the lower release rates of oxyradicals from the coated surface, nor suffers from reduced mechanical properties by the formation of voids after leaching.
Pre-exposure to ZnO-coated surfaces increases bacterial susceptibility to antibiotics
The combination of “active” antibiofilm coatings and antibiotic utilization can be a very attractive approach to reduce bacterial resistance and infection. Therefore, our next goal was to examine whether a short pre-exposure to ZnO NP-coated surfaces could enhance the susceptibility of bacteria to subsequent exposure to antibiotics. It is noteworthy that only a few publications were found dealing with investigations of the integrated treatment effect of zinc oxide with antibiotics, none of which in the form of ZnO NPs as a coating agent.34–36 In this study, bacteria suspensions were exposed for 10 min to coated glass surfaces and then plated on plates with and without antibiotics (i.e., non-selective vs. selective). The antibiotic concentration in the plates was 50% lower than the MIC for each antibiotic (i.e., sub-MIC). The comparison between the viable count that appears on the selective plate vs. the number of viable bacteria on the non-selective plate allowed us to determine whether there was any antibacterial affect. As a control, we plated bacterial suspensions that were exposed to uncoated glass surfaces. As can be seen in Fig. 6, when compared to the uncoated control sample, the viable count on the selective and non-selective plates were similar for all antibiotics. This result is, of course, not surprising, as sub-MIC concentrations were used. The short exposure to the ZnO-coated plates alone without subsequent exposure to antibiotics (i.e., plating on the non-selective plate) also did not cause substantial killing, suggesting that although radical oxygen species are produced, such a short exposure is not sufficient for killing the bacteria. However, a clear increase in bacterial susceptibility to subsequent antibiotic treatment was observed. If the counted CFU after non-selective and non–ZnO treatment is taken as 100%, then 43% reduction for E. coli in CFU was detected for Chloramphenicol (CP) and 34% for Ampicillin (AM). Other kinds of antibiotics produced a less significant difference. For the S. aureus, 19% reduction in CFU was detected for CP and 13% for AM and 10% for Erythromycin (ER), whereas other kinds of antibiotics (namely, NA and TO) did not produce a pronounced difference for the isolate. Hence, the results signify that pretreatment of ZnO coated glass increases the sensitivity of E. coli to both CP and AM. For S. aureus only a moderate enhancement of sensitivity was detected, mainly for CP.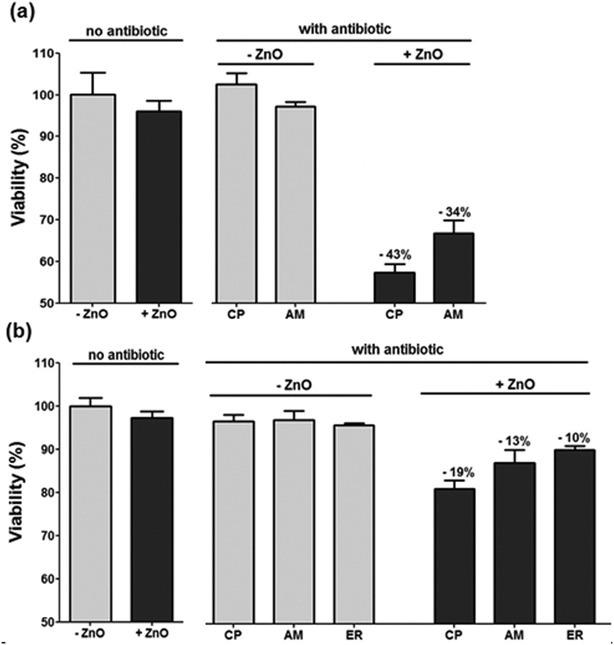 | ||
| Fig. 6 Effect of ZnO glass coating and candidate antibiotics against E. coli and S. aureus. The antibacterial effect was evaluated with non selective medium, selective media and selective media following short exposure to ZnO glass coating a) E. coli b) S. aureus. (CP: Chloramphenicol; AM: Ampicillin; ER: Erythromycin). | ||
CP functions by inhibiting the peptidyl transferase activity of the bacterial 50S subunit of the ribosome, thus preventing protein synthesis. AM inhibits the third and final stage of bacterial cell-wall synthesis. It can be hypothesized that the short pre-treatment of the bacteria with the ZnO coating induces non-lethal bacterial membrane damage, which may increase the permeation and absorption of the antibiotics into the bacterial cell, thus improving the efficacy of the antibiotics. However, the exact mechanism underlying these enhanced antibiotics actions and the reason for the difference in the antibiotics efficacies are not clear and merit further investigation.
Conclusions
Bacteria growing as a biofilm on surfaces are generally more resistant to many antimicrobial agents than the same bacteria growing in a free-swimming (planktonic) state. The resistant characteristic of biofilms leads to persistent infections in the human body, e.g., in lungs or on implant surfaces, as well as to troublesome biofilms in industrial processes that may cause malfunction of equipment. Our objective in this investigation was to probe the antibiofilm activity of a glass surface coated with ZnO NPs against two common bacterial pathogens. The antibiofilm activity of the coated film was excellent, as demonstrated by confocal microscopy. ESR measurements strongly suggest that the antibiofilm activity of ZnO NP coating is mediated through the formation of ROS. We were also able to demonstrate that even a short pre-exposure to the coated surface that by itself does not induce the substantial killing of planktonic bacteria, enhances the susceptibility of the bacteria to subsequent antibiotic treatment. Finally, several physico-chemical surface characterization methods revealed a strong adherence and deep penetration of the ZnO NPs onto/into the glass film, which was obtained by the environmentally-friendly ultrasonic method. This is explained by the driving force of the microjets, which throws the newly-formed NPs at the surface at a very high speed. Taken together, our results provide a new approach to achieve self-sterilizing properties to surfaces that can be utilized in a wide range of medical and environmental applications.References
- L. Hall-Stoodley, J. William Costerton and P. Stoodley, Nat. Rev. Microbiol., 2004, 2, 95 CrossRef CAS.
- C. A. Fux, J. W. Costerton, P. S. Stewart and P. Stoodley, Trends Microbiol., 2005, 13, 34 CrossRef CAS.
- S. Bose and A. Krishna Ghosh, J. Clin. Diagnostic Res., 2011, 5(1), 127 Search PubMed.
- D. Thomas and F. Day, Annu. Rev. Microbiol., 2007, 61, 401 CrossRef.
- R. O. Darouiche, N. Engl. J. Med., 2004, 350, 1422 CrossRef CAS.
- R. O. Darouiche, Preventing infection in surgical implants. US Surgery, 2007, 40–45 Search PubMed http://www.touchbriefings.com/pdf/2742/darouiche.pdf .
- J. Lellouche, E. Kahana, S. Elias, A. Gedanken and E. Banin, Biomaterials, 2009, 30, 5969 CrossRef CAS.
- S. Häussler and M. R. Parsek, J. Bacteriol., 2010, 192(12), 2941 CrossRef.
- A. S. Lynch and Darren Abbanat, Expert Opin. Ther. Pat., 2010, 20(10), 1373 CrossRef CAS.
- S. Makhluf, R. Dror, Y. Nitzan, Y. Abramovich, R. Jelinek and A. Gedanken, Adv. Funct. Mater., 2005, 17, 1708 CrossRef.
- G. Applerot, A. Lipovsky, R. Dror, N. Perkas, Y. Nitzan, R. Lubart and A. Gedanken, Adv. Funct. Mater., 2009, 19, 842 CrossRef CAS.
- K. R. Raghupathi, R. T. Koodali and A. C. Manna, Langmuir, 2011, 27, 4020 CrossRef CAS.
- J. Sawai, J. Microbiol. Methods, 2003, 54, 177 CrossRef CAS.
- P. K. Stoimenov, R. L. Klinger, G. L. Marchin and K. J. Klabunde, Langmuir, 2002, 18, 6679 CrossRef CAS.
- H. C. Axtell, S. M. Hartley and R. A. Sallavanti, U.S. Patent5,026,778, 2005 Search PubMed.
- H. Cao and S. L. Suib, J. Am. Chem. Soc., 1994, 116, 5334 CrossRef CAS.
- B. Ao, L. Kummerl and D. Haarer, Adv. Mater., 1995, 7, 495 CrossRef.
- M. Singhal, P. Chhabra, P. Kang and D. O. Shah, Mater. Res. Bull., 1997, 32, 239 CrossRef CAS.
- Y. Jiang, S. Decker, C. Mohs and K. J. Klabunde, J. Catal., 1998, 180, 24 CrossRef CAS.
- A. Hozumi, S. Kojima, S. Nagano, T. Seki, N. Shirahata and T. Kameyama, Langmuir, 2007, 23, 3265 CrossRef CAS.
- K. H. Lee, K. H. Huang, W. L.Tseng, T. C. Chiu, Y. W. Lin and H. T. Chang, Langmuir, 2007, 23, 1435 CrossRef CAS.
- R. Gottesman, S. Shukla, N. Perkas, L. A. Solovyov, Y. Nitzan and A. Gedanken, Langmuir, 2011, 27(2), 720 CrossRef CAS.
- Y. Didenko and K. S. Suslick, Nature, 2002, 418, 394 CrossRef CAS.
- A. Gedanken, Ultrason. Sonochem., 2004, 11(2), 47 CrossRef CAS.
- K. Ghule, A. V. Ghule, B. J. Chen and Y. C Ling, Green Chem., 2006, 8, 1034 RSC.
- G. Applerot, N. Perkas, G. Amirian, O. Girshevitz and A. Gedanken, Appl. Surf. Sci., 2009, 256S, S3 CrossRef.
- I. Perelshtein, G. Applerot, N. Perkas, E. Wehrschetz-Sigl, A. Hasmann, G. M. Guebitz and A. Gedanken, ACS Appl. Mater. Interfaces, 2009, 1(2), 361 CAS.
- Y. Nitzan and M. Kauffman, Lasers Med. Sci., 1999, 14, 269 CrossRef.
- A. Kotlyar, N. Perkas, Y. Koltypin, M. Meyer, W. Zimmermann and A. Gedanken, Ultrason. Sonochem., 2008, 15, 839 CrossRef.
- M. B. H. Breese, G. W. Grime and F. Watt, Annu. Rev. Nucl. Part. Sci., 1992, 42, 1 CrossRef CAS.
- A. Lipovsky, Z. Tzitrinovich, H. Friedmann, G. Applerot, A. Gedanken and R. Lubart, J. Phys. Chem. C, 2009, 113, 15997 CAS.
- N. Asakuma, T. Fukui, M. Toki, K. Awazu and H. Imai, Thin Solid Films, 2003, 445, 284 CrossRef CAS.
- G. Sengupta, H. S. Ahluwalia, S. Banerjee and S. P. Sen, J. Colloid Interface Sci., 1979, 69, 217 CrossRef CAS.
- J. Sawai, H. Kojima, H. Igarashi, A. Hashimoto, S. Shoji, T. Kokugan, M. Shimizu and H. Kojima, J. Ferment. Bioeng., 1998, 86, 521 CrossRef CAS.
- V. Thati, A. S. Roy, M. V. N. Ambika Prasad, C. T. Shivannavar and S. M. Gaddad, J. Biosci. Tech., 2010, 1(2), 64 Search PubMed.
- M. Banoee, S. Seif, Z. E. Nazari, P. Jafari-Fesharaki, H. R. Shahverdi, A. Moballegh, K. M. Moghaddam and A. R. Shahverdi, J. Biomed. Mater. Res. B. Appl. Biomater., 2010, 93(2), 557 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra00602b/ |
| This journal is © The Royal Society of Chemistry 2012 |