Luminescence of oxyfluoride glasses co-doped with Ag nanoclusters and Yb3+ ions
V. K.
Tikhomirov
*a,
T.
Vosch
b,
E.
Fron
c,
V. D.
Rodríguez
d,
J. J.
Velázquez
d,
D.
Kirilenko
e,
G.
Van Tendeloo
e,
J.
Hofkens
c,
M.
Van der Auweraer
c and
V. V.
Moshchalkov
a
aINPAC-Institute for Nanoscale Physics and Chemistry, Katholieke Universiteit Leuven, Belgium. E-mail: Victor.Tikhomirov@fys.kuleuven.be
bNano-Science Center, Department of Chemistry, University of Copenhagen, Universitetsparken, 5, 2100, Copenhagen, Denmark
cDepartment of Chemistry, Katholieke Universiteit Leuven, Belgium
dDepartamento de Física Fundamental y Experimental, Electrónica y Sistemas, Universidad de La Laguna, Tenerife, Spain
eEMAT, Electron Microscopy for Materials Science, Antwerpen Universiteit, Belgium
First published on 19th December 2011
Abstract
Bulk oxyfluoride glasses co-doped with Ag nanoclusters and Yb3+ ions have been prepared by a melt quenching technique. When excited in the absorption band of the Ag nanoclusters between 300 to 500 nm, these glasses emit a broad band characteristic of the Ag nanoclusters between 400 to 750 nm as well as an emission band between 900 to 1100 nm, originating from Yb3+ ions. The intensity ratio of the Yb3+/Ag emission bands increases with the Ag doping level at a fixed concentration of Yb3+, indicating the presence of energy transfer mechanism from the Ag nanoclusters to the Yb3+ ions. Comparison of time-resolved decay kinetics of the luminescence in the respectively Ag nanocluster-Yb3+ co-doped and single Ag nanocluster doped glasses, hints towards an energy transfer from the red and infrared emitting Ag nanoclusters to the Yb3+ ions.
1. Introduction
Photoluminescence of silver (Ag) nanoclusters has recently attracted a major interest due to promising applications as optical nanolabels, nanosensors, in optical recording, as visible light sources, etc., see ref. 1–23 and references therein. It has also been reported that larger Ag nanoparticles, typically of a size about 5 to 10 nm, when embedded in a glassy host, can amplify the luminescence of rare-earth dopants in that host, i.e. of Eu3+ or Er3+, e.g. in ref. 24 and 25 and references therein. The amplification is supposed to occur due to plasmon oscillations in the Ag nanoparticles resulting in local gradients of the electric field and an increase of the probability of the optical transitions in the rare-earth dopants, e.g. in ref. 24–27. Synthetic silver metal nano-patterns can also amplify the emission of luminescent nanoparticles.26,27Here, we report on the preparation of oxyfluoride glass co-doped with Ag nanoclusters and rare-earth ions Yb3+. The Ag nanoclusters are spontaneously created in this glass on casting without further heat treatment steps. While the Ag nanoclusters in this glass do emit themselves in the visible, they in addition, transfer some part of the excitation energy to the Yb3+ co-dopants, which then emit in the near infrared. These phenomena may have applications in UV-driven light sources and lasers, and solar spectrum down-conversion for enhancement the efficiency of Si solar cells, e.g. in ref. 28.
2. Experimental
The procedure for the preparation of oxyfluoride glass doped with rare-earth ions, such as Yb3+, 32(SiO2)9(AlO1.5)31.5(CdF2)18.5(PbF2)5.5(ZnF2):3.5(YbF3), mol%, has been described in ref. 29,30. Here we have followed that procedure but, in addition, have also added AgNO3, when preparing the batch. The 3.5 mol% of YbF3 is an equivalent of 6.19 wt% of YbF3 and further in the text the doping level by YbF3 will be addressed in wt% for easier comparison with the doping level by AgNO3, which is provided also in wt%. Higher doping with YbF3 decreases the glass stability of these oxyfluoride glasses29 and therefore it was not undertaken. The glass melt was cast into the mold preheated to 300 °C and then allowed to cool down in the air. No further deliberate heat-treatment steps were undertaken in preparation of the glass samples studied in this work. The resulting pieces of glass, normally of 4 × 1 × 0.3 cm3 in size, were then polished and cut in smaller pieces for optical and structural characterization.The steady state spectra have been recorded using conventional Photon Technology and SPEX spectrofluorimeters equipped with Xenon CW-lamps, UV/VIS photomultiplier tubes (250–750 nm sensitivity range), and a NIR detector (cooled InGaAs detector, 600–1700 nm sensitivity range). The samples have been measured in back reflection geometry to reduce self-absorption. The spectral response functions of the spectrometers have been calibrated with standard light sources and spectra were recorded for this response.
Luminescence decay times in the pico and nanosecond time scales have been measured by means of a time-correlated single photon counting (TC-SPC) setup described previously.31 The repetition rate of the excitation pulses was 8.1 MHz. The decays were recorded with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 counts in the peak channel in a time window of 9 ns corresponding to 2.2 ps/ch, and analyzed globally with in house developed time resolved fluorescence analysis (TRFA) software.32 The full width at half-maximum (FWHM) of the instrument response function was typically in the order of 40 ps. The quality of the fits has been judged by the fit parameters χ2 (< 1.2), Zχ2 (< 3) and the Durbin Watson parameter (1.8 < DW < 2.2) as well as by the visual inspection of the residuals and autocorrelation function.32
000 counts in the peak channel in a time window of 9 ns corresponding to 2.2 ps/ch, and analyzed globally with in house developed time resolved fluorescence analysis (TRFA) software.32 The full width at half-maximum (FWHM) of the instrument response function was typically in the order of 40 ps. The quality of the fits has been judged by the fit parameters χ2 (< 1.2), Zχ2 (< 3) and the Durbin Watson parameter (1.8 < DW < 2.2) as well as by the visual inspection of the residuals and autocorrelation function.32
Luminescence decay times in the microsecond time scale have been recorded with a digital store oscilloscope. The repetition rate of the 10 ns excitation pulses was 20 Hz; they were generated by optical parametric oscillator (OPO) pumped by a third harmonic of YAG:Nd laser beam.
Transmission electron microscopy (TEM) studies have been performed on a JEOL JEM-3000F microscope (0.19 nm point-to-point resolution) equipped with a GIF-2000 spectrometer. Elemental maps for Yb and Ag atoms have been obtained by means of energy filtered TEM (EFTEM) using Yb-N4,5 and Ag-M4,5 inner-shell edges in the electron energy loss spectrum (EELS) of the samples.
3. Results and discussion
3.1 Steady state luminescence
When doping the oxyfluoride glass with AgNO3, a green/yellow luminescence is observed in the glass samples upon UV excitation. The latter is also the case when the oxyfluoride glass is co-doped with AgNO3 and Yb3+. Fig. 1(a) shows a daylight picture of three glass samples co-doped with 1, 3 and 9 wt% of AgNO3 each containing also 6.19 wt% of YbF3. The glass pieces are transparent and slightly gold colored. Upon holding the glass samples under a UV lamp, green/yellow emission can be clearly observed from these samples, Fig. 1(b), while the intensity of the emission increases with the AgNO3 content.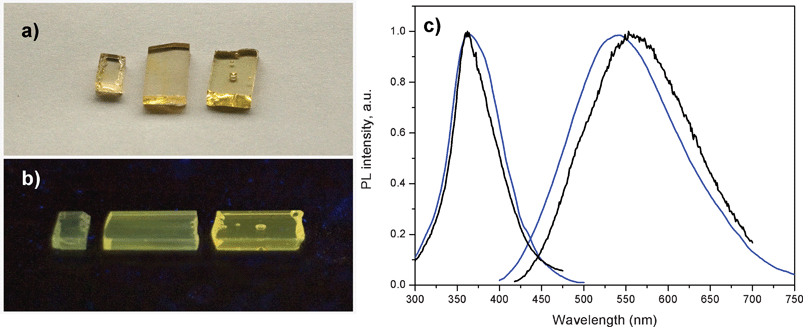 | ||
| Fig. 1 a) Daylight picture of glass samples containing from left to right 1, 3 and 9 wt% of AgNO3. All three samples also contain 6.19 wt% of YbF3. b) Luminescence image of the same glass samples excited with a UV lamp at 366 nm. c) Excitation and emission spectra of a glass samples containing 1 wt% of AgNO3 and no YbF3 (black curves) and a glass sample containing 1 wt% of AgNO3 and 6.19 wt% of YbF3 (blue curves). Excitation was at 380 nm for the emission spectra, and emission was collected at 550 nm for the excitation spectra. | ||
Excitation and emission spectra of the glass sample co-doped with 1 wt% of AgNO3 and 6.19 wt% of YbF3, and of glass sample single doped with 1 wt% of AgNO3, are shown in Fig. 1(c). The emission seen in Fig. 1(c) clearly originates from the silver dopants in the glass samples, since similar prepared glass samples containing no silver nitrate but either 6.19 wt% or 0 wt% of YbF3 dopant, showed no green/yellow emission at the same UV excitation (data not shown). The excitation and emission spectra in Fig. 1(c) look similar, indicating that they originate from silver related species. The blue shift of luminescence in the samples containing YbF3 is likely related to preferential quenching (energy transfer) of red-emitting Ag nanoclusters by the Yb3+, as it will be shown in Time Resolved Luminescence Section.
Fig. 2 shows the normalized excitation and emission spectra of the Ag nanoclusters in glass samples containing 1 wt% of AgNO3 and 6.19 wt% of YbF3. Upon blue shifting of the excitation wavelength (from 460 to 310 nm), the maximum of Ag nanoclusters emission also does shift to the blue (from 580 to 470 nm).
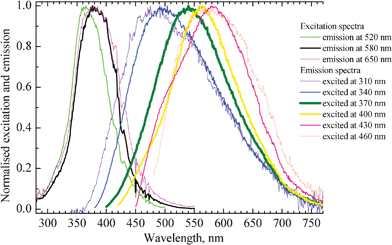 | ||
| Fig. 2 Excitation and emission spectra of a glass sample co-doped with 1 wt% of AgNO3 and 6.19 wt% of YbF3. Excitation and emission wavelengths of the corresponding curves are indicated on the right side of the figure. Thickening of the lines corresponds to strengthening of intensity of emission and excitation, respectively. | ||
The thickening of lines in Fig. 2 corresponds to strengthening of the emission and excitation intensity, respectively. The strongest emission occurs in the green-yellow part of the spectrum from about 540 to 570 nm and this can be most efficiently observed when excitation wavelengths from 370 to 380 nm are used. The position and shape of the emission and excitation spectra match well with the earlier reported respective spectra of Ag nanoclusters dispersed in zeolites.4,33 The red shift of the emission spectrum upon increasing the excitation wavelength suggests that a distribution of silver clusters with different size and geometry is present in the glass.
While Fig. 1 and 2 represent the emission properties of the Ag nanoclusters, Fig. 3 and 4 show that the photoexcited Ag nanoclusters also transfer some of their excitation energy to the Yb3+ co-dopants. Fig. 3 shows the full emission spectra of the co-doped glass samples when excited at 380 nm into the excitation band of the Ag nanoclusters, (see Fig. 1 and 2). The emission bands from both the Ag nanoclusters (peaking at about 560 nm) and the Yb3+ ions (peaking at about 1000 nm) can be seen, while the ratio of the Yb3+/Ag bands increases substantially with AgNO3 doping when the Yb3+ concentration remains the same. This indicates the presence of a mechanism that transfers the excited state energy of the Ag nanoclusters to the Yb3+ ions. Although the relative luminescence intensity between different samples in Fig. 3 may be affected by external factors, such as sample shape and size, orientation of samples with respect to excitation and detection optical paths, the ratio between emission bands of the Ag nanoclusters and Yb3+ ions is ensured by the calibrated spectral response of the setup. A minor self-absorption effect at the 975 nm shoulder of the Yb3+ band may be present, while the 1020 nm shoulder of the Yb3+ band is not affected by self-absorption because Yb3+ does not absorb at this wavelength in this host, see Fig. 2 in ref. 34.
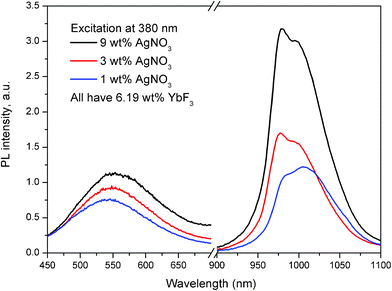 | ||
| Fig. 3 Emission spectra of glass samples containing 1, 3 and 9 wt% of AgNO3 and 6.19 wt% of YbF3. The glass samples were excited at 380 nm and the Ag nanocluster emission and Yb3+ emission were rescaled to correct for the detectors sensitivities. | ||
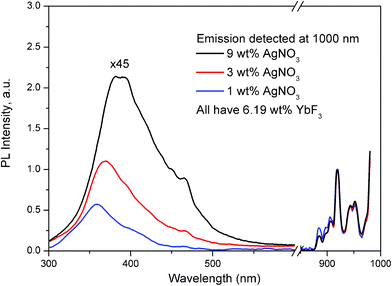 | ||
| Fig. 4 Excitation spectra of Yb3+ dopant in glass samples containing 1, 3 and 9 wt% of AgNO3 and 6.19 wt% of YbF3, detecting the emission at 1000 nm. The spectra were normalized to the Yb3+ dopant emission at 925 nm. For convenient viewing, the intensity of the spectrum from 300 until 700 nm for all three curves was multiplied by a factor of 45. | ||
It should also be noted when exciting a glass containing 6.19 wt% of YbF3 and no AgNO3 at 380 nm, also emission from the Yb3+ ions was observed (data not shown). The amount of Yb3+ emission, in this case, corresponded to about 1/3 of the emission observed in the glass sample containing 9 wt% of AgNO3 and 6.19 wt% of YbF3. The latter indicates that in the glass containing only 6.19 wt% of YbF3 another pathway of energy transfer to Yb3+ ions from the host is present; an effect that has been previously described in the literature, ref. 35 and references therein.
Fig. 4 shows excitation spectra of Yb3+ dopant, when emission of the Yb3+ was measured at 1000 nm. The left part of Fig. 4 corresponds to excitation viaAg nanoclusters, and the right part corresponds to the direct excitation of the Yb3+ ions. The excitation spectra were normalized to the Yb3+ dopant excitation peak at 925 nm because all three glass samples contain the same amount of Yb3+ dopant. We can clearly see that the Ag related part of the excitation spectrum (from 300 until 500 nm), increases systematically with increasing Ag doping level from 1 until 9 wt% AgNO3. Also, the excitation band of the emission from Yb3+ for the sample co-doped with 1 wt% AgNO3 (blue curve in Fig. 4) matches well the excitation band of Ag nanoclusters in the respectively 1 wt% single AgNO3 doped sample in Fig. 1(c). These data indicate the presence of an excitation energy transfer from Ag nanoclusters to Yb3+ dopants.
3.2. Time-resolved luminescence
Time-correlated single photon experiments with pico- and nanosecond time resolution were performed on the luminescence of the Ag nanoclusters in samples single doped with AgNO3 and co-doped with AgNO3 and YbF3. The samples were excited at 375 nm. The decay curves were detected at 500, 550, 600 and 650 nm and were globally analyzed, as described in ref. 32. Fig. 5 shows an example of decay curves recorded at 550 nm emission.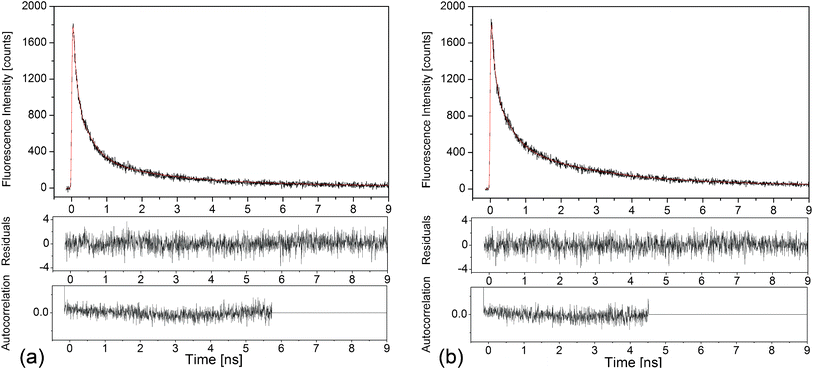 | ||
| Fig. 5 Time-correlated single photon counting measurement of the luminescence decay kinetics at 550 nm of the Ag nanoclusters (black curves), and the corresponding fits, red curves, for (a) glass doped with 9 wt% AgNO3 and (b) glass co-doped with 9 wt% AgNO3 and 6.19 wt% YbF3. Residuals and autocorrelation traces are shown in the lower panels, respectively: χ2 = 1.065 in (a) and χ2 = 1.100 in (b). | ||
The respective decay curves were found to be well fit with a four-exponential function and the resulting amplitudes of all four decay parameters are shown in Fig. 6 as a function of emission wavelength. As can be seen from Fig. 6, no major differences are observed in the amplitudes and values of the respective decay components for the studied samples. Both samples can be globally fitted with four exponential values around 50 ps, 300 ps, 1600 ps and a longer component around 7.5 ns. These results indicate that the emission of the Ag clusters that emit in the emission range from 500 nm to 600 nm is not significantly quenched when co-doped with Yb3+ ions. This is not surprising since the energy gap between the emission of the Ag nanoclusters, which emit between 500 to 600 nm, and the absorption of the Yb3+ ions at 900 nm is fairly large for a Förster or Dexter type energy transfer mechanism to occur.
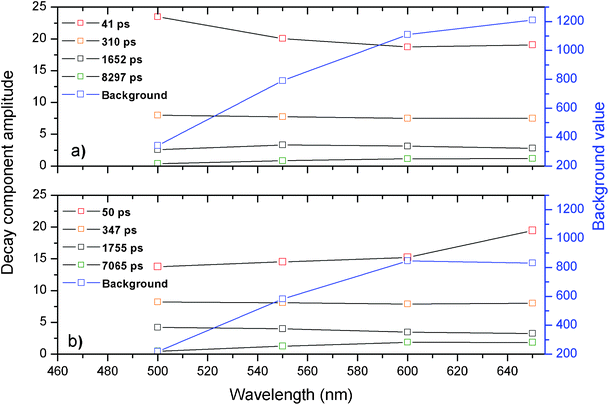 | ||
| Fig. 6 Overview of the amplitudes from the decay time components as a function of emission wavelength for a) glass doped with 9 wt% AgNO3 and b) glass co-doped with 9 wt% AgNO3 and 6.19 wt% YbF3. The black curves are the amplitudes corresponding to the globally fitted decay times. The blue curve corresponds to the intensity of the constant background that was present in the decay curve. Decay curves were recorded until the 10000 counts in the maximum. | ||
The multi-exponential decay in the 500 nm to 600 nm wavelength range agrees well with decay time values reported for yellow emitting Ag nanoclusters dispersed in zeolite matrices.4 However, in the zeolite work,4 it was also shown that the red emitting Ag nanoclusters may be formed which have a luminescence decay time in the tens to hundreds of microsecond range.4
When fitting the luminescence decay curves of the Ag nanoclusters in these glass samples, we observed a substantial increase of a constant background level with increasing the detection wavelength. The value of the constant background level has also been plotted by a blue curve in Fig. 6. The increase of this constant background level upon increasing emission wavelength can be explained by assuming that the red emitting Ag nanoclusters were formed in these glasses, which have decay times in the microsecond time scale region, similar as it was observed in the zeolite matrices.4
From the fitted decay curves, we can observe that the constant background level, at 650 nm, decreases for the sample containing Yb3+ while it further increases for the sample containing no Yb3+ ions. This observation hints that the red/near infrared emitting Ag nanoclusters are responsible for the energy transfer to the Yb3+ ions. A relative increase of short-lived 50 ps decay component with addition of Yb3+ dopants observed at 650 nm also hints to energy transfer from the red/near infrared emitting Ag nanoclusters to the Yb3+ co-dopants.
Having such hints, we have measured the microsecond time scale decay kinetics in the single Ag nanoclusters doped and Ag nanoclusters-Yb3+ co-doped glass samples, when detecting the decays at red and infrared wavelengths; an example is shown in Fig. 7 when detection was at 800 nm. Here the excitation was done at 420 nm because excitation at this wavelength produces a substantial emission of Ag nanoclusters in the red and near infrared parts of the spectrum, e.g. in Fig. 2.
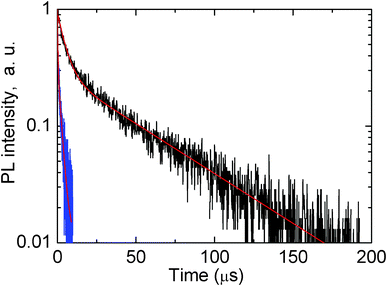 | ||
| Fig. 7 Logarithm decay kinetics of luminescence of glass doped with 5 wt% AgNO3 (black curve) and glass co-doped with 5 wt% AgNO3 and 6.19 wt% YbF3 (blue curve). Detection of decays was at 800 nm, excitation of luminescence was at 420 nm. Red curves show two-exponential fits. | ||
It is clearly seen in Fig. 7 that co-doping with Yb3+ greatly quenches the red/near infrared emission of Ag nanoclusters due to an efficient energy transfer from the Ag nanoclusters emitting in the red/infrared to the Yb3+ co-dopants, in agreement with suggestions made above in the text.
3.3. Transmission electron microscopy
Fig. 8 shows an energy filtered transmission electron microscope (EFTEM) image of a typical fragment of an Ag nanocluster and Yb3+ co-doped glass sample. EFTEM allows mapping of the distribution of a particular chemical element by filtering electrons of particular energy loss corresponding to the characteristic inner-shell transition of that element. Fortunately, both Ag and Yb have suitable edges in the spectrum of this glass, and therefore the electron energy loss spectrum from these elements was used for mapping. Tiny, about 1 nm and smaller, Ag nanoclusters are seen distributed randomly throughout Fig. 8 while a large number of them are closer grouped in the centre of this image. The Yb3+ dopant shows also fluctuations in the distribution throughout the glass; clearly a higher concentration can be observed next to the central conglomerate of Ag nanoclusters, while less Yb3+ is present at the left upper and bottom corners. The image in Fig. 8 confirms the presence of Ag nanoclusters in this glass, and the possibility for energy transfer from the Ag nanoclusters to the Yb3+ dopants is feasible due to very short distances between them.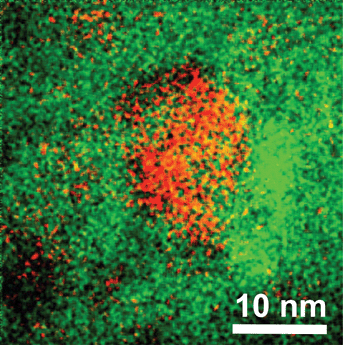 | ||
| Fig. 8 Energy filtered transmission electron microscope image of the piece of as-prepared glass co-doped with Ag nanoclusters and Yb3+ ions; red color represents Ag and green color Yb atoms, respectively. | ||
The distribution of distances between the Ag nanoclusters and Yb3+ dopants may be investigated bearing in mind some degree of 3-dimensionality of picture presented in Fig. 8. However, new software is to be developed for this which we plan to do in the future.
The mechanism for the formation of Ag nanoclusters in this glass will be addressed in an extensive future work, which is to deal with computation methods of quantum chemistry such as molecular orbitals and perturbation theory. Here we note that the presence of fluoride components of PbF2 and CdF2 in these oxyfluoride glasses seems to be crucial for dispersion/dissolution of the Ag nanoclusters in the glass network; we found that these glasses without the fluoride components do not dissolve Ag nanoclusters but instead the silver forms macro-crystalline agglomerations on melting and casting the glass. We assume that the high superionic conductivity of Pb–Cd–F and Ag–F components36 ensure creation and dispersion of Ag nanoclusters in these oxyfluoride glasses.
Conclusions
An oxyfluoride glass co-doped with Ag nanoclusters and Yb3+ ions has been prepared. The glassy state of this material hints to possibility for its preparation in film and fiber form. When optically excited in the UV-blue absorption band of the Ag nanoclusters, this glass emits both the visible band of the Ag nanoclusters and the near-infrared band of the Yb3+ dopants. The effect has a potential for application in very broad band light sources and lasers. It may be also used for light frequency down-conversion applications.28,37Acknowledgements
Financial support of the “Fonds voor Wetenschappelijk Onderzoek FWO” (Grants G.0402.09 and G.0181.10), the K.U. Leuven Research Fund (GOA 2004/2 and 2006/2), Inter-Disciplinair Onderzoek (IDO/07/011), the Flemish Government (long term structural funding- Methusalem funding CASAS), the Federal Science Policy of Belgium (IAP-VI/27), the Hercules foundation (HER/08/021) and the ‘Industrieel Onderzoeksfonds (IOF/07/HB023 and IOF/09/HB044) is gratefully acknowledged. E.F. thanks the K.U.Leuven Research Fund for a postdoctoral grant. T.V. thanks the UNIK Synthetic Biology program (Grant 09–065274) for financial support. We are grateful to Ministerio de Ciencia e Innovación for financial support (MAT2009-12079) and Gobierno Autónomo de Canarias: Agencia Canaria de Investigación, Innovación y Sociedad de la Información (Sol-SubC200801000286).References
- L. A. Peyser, A. E. Vinson, A. P. Bartko and R. M. Dickson, Science, 2001, 291, 103 CrossRef CAS.
- G. de Cremer, B. F. Sels, J. Hotta, M. E. Bartholomeeusen, E. Coutiño-Gonzales, D. E. De Vos, T. Vosch and J. Hofkens, Adv. Mater., 2010, 22, 957 CAS.
- T. Vosch, Y. Antoku, J. C. Hsiang, C. I. Richards, J. I. Gonzalez and R. M. Dickson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12616 CrossRef CAS.
- G. De Cremer, E. Coutiño-Gonzalez, M. B. J. Roeffaers, B. Moens, J. Ollevier, M. Van der Auweraer, R. Schoonheydt, P. A. Jacobs, F. C. De Schryver, J. Hofkens, D. E. De Vos, B. F. Sels and T. Vosch, J. Am. Chem. Soc., 2009, 131, 3049 CrossRef CAS.
- G. De Cremer, Y. Antoku, M. B. J. Roeffaers, M. Sliwa, J. Van Noyen, S. Smout, J. Hofkens, D. E. De Vos, B. F. Sels and T. Vosch, Angew. Chem., Int. Ed., 2008, 47, 2813 CrossRef CAS.
- L. Maretti, P. S. Billone, Y. Liu and J. C. Scaiano, J. Am. Chem. Soc., 2009, 131, 13972 CrossRef CAS.
- Z. Shen, H. Duan and H. Frey, Adv. Mater., 2007, 19, 349 CrossRef CAS.
- I. Díez, M. Pusa, S. Kulmala, H. Jiang, A. Walther, A. S. Goldman, A. Müller, O. Ikkala and R. Ras, Angew. Chem., Int. Ed., 2009, 48, 2122 CrossRef CAS.
- C. I. Richards, S. Choi, J. C. Hsiang, Y. Antoku, T. Vosch, A. Bongiorno, Y. L. Tzeng and R. M. Dickson, J. Am. Chem. Soc., 2008, 130, 5038 CrossRef CAS.
- C. I. Richards, J. C. Hsiang, D. Senapati, S. Patel, J. Yu, T. Vosch and R. M. Dickson, J. Am. Chem. Soc., 2009, 131, 4619 CrossRef CAS.
- S. A. Patel, M. Cozzuol, J. M. Hales, C. I. Richards, M. Sartin, J. C. Hsiang, T. Vosch, J. W. Perry and R. M. Dickson, J. Phys. Chem. C, 2009, 113, 20264 CrossRef CAS.
- Y. Antoku, J.-i. Hotta, H. Mizuno, R. M. Dickson, J. Hofkens and T. Vosch, Photochem. Photobiol. Sci., 2010, 9, 716 RSC.
- W. W. Guo, J. P. Yuan, Q. Z. Dong and E. K. Wang, J. Am. Chem. Soc., 2010, 132, 932 CrossRef CAS.
- J. Yu, S. Choi and R. M. Dickson, Angew. Chem., Int. Ed., 2009, 48, 318 CrossRef CAS.
- L. H. Xiao, Y. He and E. S. Yeung, J. Phys. Chem. C, 2009, 113, 5991 Search PubMed.
- Z. K. Wu, E. Lanni, W. Q. Chen, M. E. Bier, D. Ly and R. C. Jin, J. Am. Chem. Soc., 2009, 131, 16672 CrossRef CAS.
- L. Shang and S. J. Dong, Biosens. Bioelectron., 2009, 24, 1569 CrossRef CAS.
- B. Sengapta, K. Springer, J. G. Buckman, S. P. Story, O. H. Abe, Z. W. Hasan, Z. D. Prudowsky, S. E. Rudisill, N. N. Degtyareva and J. T. Petty, J. Phys. Chem. C, 2009, 113, 19518 CrossRef CAS.
- P. R. O'Neil, L. R. Velazquez, D. G. Dunn, E. G. Gwinn and D. K. Fygenson, J. Phys. Chem. C, 2009, 113, 4229 CrossRef CAS.
- K. V. Mrudula, T. U. B. Rao and T. Pradeep, J. Mater. Chem., 2009, 19, 4335 RSC.
- W. W. Guo, J. P. Yuan and E. K. Wang, Chem. Commun., 2009, 23, 3395 Search PubMed.
- J. Yu, S. Choi, C. I. Richards, Y. Antoku and R. M. Dickson, Photochem. Photobiol., 2008, 84, 1435 CrossRef CAS.
- B. Sengapta, C. M. Ritchie, J. G. Buckman, K. R. Johnsen, P. M. Goodwin and J. T. Petty, J. Phys. Chem. C, 2008, 112, 18776 CAS.
- O. L. Malta, P. A. Santa-Cruz, G. F. de Sa and F. Auzel, J. Lumin., 1985, 33, 261 Search PubMed.
- L. R. P. Kassab, F. A. Bomfim, J. R. Martinelli, N. I. Wetter, J. J. Neto and C. D. de Araújo, Appl. Phys. B: Lasers Opt., 2009, 94, 239 Search PubMed.
- P. P. Pompa, L. Martiradonna, A. Della Torre, F. Della Sala, L. Manna, M. De Vittorio, F. Calabi, R. Cingolani and R. Rinaldi, Nat. Nanotechnol., 2006, 1, 126 CrossRef CAS.
- P. Lalanne, J. P. Hugonin, G. Juliè, V. Mathet and M. Mortier, Phys. Rev. Lett., 2007, 98, 153902 CrossRef CAS.
- V. D. Rodríquez, V. K. Tikhomirov, J. Mendez-Ramos, A. C. Yanes and V. V. Moshchalkov, Sol. Energy Mater. Sol. Cells, 2010, 94, 1612 Search PubMed.
- V. K. Tikhomirov, D. Furniss, I. M. Reaney, M. Beggiora, M. Ferrari, M. Montagna and R. Rolli, Appl. Phys. Lett., 2002, 81, 1937 CrossRef CAS.
- V. K. Tikhomirov, L. F. Chibotaru, D. Saurel, P. Gredin, M. Mortier and V. V. Moshchalkov, Nano Lett., 2009, 9, 721 Search PubMed.
- M. Maus, E. Rousseau, M. Cotlet, G. Schweitzer, J. Hofkens, M. Van der Auweraer and F. C. De Schryver, Rev. Sci. Instrum., 2001, 72, 36 CrossRef CAS.
- N. Boens, W. W. Qin, N. Basaric, J. Hofkens, M. Ameloot, J. Pouget, J. P. Lefevre, B. Valeur, E. Gratton, M. Vandeven, N. D. Silva, Y. Engelborghs, K. Willaert, A. Sillen, G. Rumbles, D. Phillips, A. Visser, A. van Hoek, J. R. Lakowicz, H. Malak, I. Gryczynski, A. G. Szabo, D. T. Krajcarski, N. Tamai and A. Miura, Anal. Chem., 2007, 79, 2137 CrossRef CAS.
- G. De Cremer, E. Coutiño-Gonzalez, M. B. J. Roeffaers, D. E. De Vos, J. Hofkens, T. Vosch and B. F. Sels, ChemPhysChem, 2010, 11, 1627 CAS.
- V. K. Tikhomirov, K. Driesen, C. Görller-Walrand and M. Mortier, Opt. Express, 2007, 15, 9535 CrossRef CAS.
- S. Comby, F. Gumy, J.-C. Bünzli, T. Saraidarov and R. Reisfeld, Chem. Phys. Lett., 2006, 432, 128 CrossRef CAS.
- S. Hull, Rep. Prog. Phys., 2004, 67(1233), 1241 Search PubMed.
- B. M. van der Ende, L. Aarts and A. Meijerink, Adv. Mater., 2009, 21, 3073 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |