DOI:
10.1039/C1RA00897H
(Paper)
RSC Adv., 2012,
2, 1404-1415
Transition of trigonal-bipyramidal to square-planar geometries by oxidation of a coordinated tripodal polyphosphine in Pd(II) and Pt(II) complexes†
Received
14th October 2011
, Accepted 26th October 2011
First published on 16th December 2011
Abstract
The oxidation reactions of the unusual d8 trigonal-bipyramidal systems [MX(PP3)]X (M = Pd: X = Cl(1), Br(2), I(3); M = Pt: X = Cl(4), Br(5), I(6); PP3 = tris[2-(diphenylphosphino)ethyl]phosphine) by dissociation of the bound phosphino groups with H2O2 in chloroform and with DMSO-d6 in the presence of reduced glutathione (GSH) have been explored with emphasis on the factors that influence the number of M–X and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functionalities of the resulting products. The oxidations with H2O2 required a higher concentration for Pd than Pt and occurred with formation of both ionic distorted square-planar compounds [MX(PP2PO)]X [M = Pd: X = Cl(7); M = Pt: X = Cl(10), Br(11)], that contain one dangling PO group, and the less common neutral species [MX2(PP(PO)2] [M = Pd: X = Br(8), M = Pt: X = I(12)] showing MX2P2 environments and two PO arms, or with release of all bound phosphorus from 3 to give OP(PO)3 (9). Although there was no evidence of the formation of neutral species with two Pt–Cl bonds and dissociation of one bound phosphorus by adding H2O2 or M′Cl (M′ = Cu, Ag) to 10, the interaction with [AuCl(tdg)] (tdg = thiodiglycol) afforded [PtAuCl3(PP2PO)] (10a) containing dangling PO and PAuCl arms and one chiral central phosphorus. Thus, while the halide anions and ligands allowed both ionic (7, 10, 11) and neutral (8, 9, 10a, 12) systems, the larger anions [S(CH3)3(CH2)3(SO3)]− or GS− favour only the ionic compounds, namely [PdCl(PP2PO)][S(CH3)3(CH2)3(SO3)] (7a), obtained in coexistence with [Pd(GS)(PP2PO)]Cl (7a′) from the reaction of 1 + 3 eq. GSH in DMSO-d6. The X-ray structure of 7a confirmed the distorted ClPdP3 arrangement and the presence of a dangling PO group predicted by LSI-MS and 31P{1H}NMR data.
O functionalities of the resulting products. The oxidations with H2O2 required a higher concentration for Pd than Pt and occurred with formation of both ionic distorted square-planar compounds [MX(PP2PO)]X [M = Pd: X = Cl(7); M = Pt: X = Cl(10), Br(11)], that contain one dangling PO group, and the less common neutral species [MX2(PP(PO)2] [M = Pd: X = Br(8), M = Pt: X = I(12)] showing MX2P2 environments and two PO arms, or with release of all bound phosphorus from 3 to give OP(PO)3 (9). Although there was no evidence of the formation of neutral species with two Pt–Cl bonds and dissociation of one bound phosphorus by adding H2O2 or M′Cl (M′ = Cu, Ag) to 10, the interaction with [AuCl(tdg)] (tdg = thiodiglycol) afforded [PtAuCl3(PP2PO)] (10a) containing dangling PO and PAuCl arms and one chiral central phosphorus. Thus, while the halide anions and ligands allowed both ionic (7, 10, 11) and neutral (8, 9, 10a, 12) systems, the larger anions [S(CH3)3(CH2)3(SO3)]− or GS− favour only the ionic compounds, namely [PdCl(PP2PO)][S(CH3)3(CH2)3(SO3)] (7a), obtained in coexistence with [Pd(GS)(PP2PO)]Cl (7a′) from the reaction of 1 + 3 eq. GSH in DMSO-d6. The X-ray structure of 7a confirmed the distorted ClPdP3 arrangement and the presence of a dangling PO group predicted by LSI-MS and 31P{1H}NMR data.
Introduction
The chemical modifications of phosphines1–6 to afford phosphine oxides6–32 is an important aim of synthetic studies because of their various applications in catalysis and medicine, the preparation of mixed-metal assemblies or polymers, the use as extractive agents or carriers and the construction of nanoparticles, depending on the number of PO functionalities.8,9,24–32 In previous work, we have described the ligand exchange and selective phosphine oxidation of the distorted square-planar Pd(II)–triphos complex [PdBr(triphos)]Br (triphos = bis(2-diphenylphosphinoethyl)phenylphosphine)14e to give the ionic system [Pd(triphos-O)2][PdBr4] with the square-planar cation showing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 Pd to ligand ratio and two dangling PO arms. Recently, we confirmed that PP3 produces dissociation of two bound phosphino groups of the uncommon trigonal-bipyramidal d8 complexes [MX(PP3)]X (M = Pd, Pt; X = Cl, Br, I)33 leading to [M(PP3)2]X2 (X = Cl, Br) that show an MP4 square-planar arrangement and four dangling phosphorus available for oxidation. With the aim of increasing the knowledge about oxidations of coordinated phosphines, in this paper we report studies on the stepwise selective oxidation with H2O2 for [MX(PP3)]X and their halo-ligand/counter anion substitution reactions with glutathionate/S(CH3)3–propane sulfonato.
:2 Pd to ligand ratio and two dangling PO arms. Recently, we confirmed that PP3 produces dissociation of two bound phosphino groups of the uncommon trigonal-bipyramidal d8 complexes [MX(PP3)]X (M = Pd, Pt; X = Cl, Br, I)33 leading to [M(PP3)2]X2 (X = Cl, Br) that show an MP4 square-planar arrangement and four dangling phosphorus available for oxidation. With the aim of increasing the knowledge about oxidations of coordinated phosphines, in this paper we report studies on the stepwise selective oxidation with H2O2 for [MX(PP3)]X and their halo-ligand/counter anion substitution reactions with glutathionate/S(CH3)3–propane sulfonato.
Results and discussion
The use of donor solvents was restricted to the reactions involving species with low solubility in the common non-coordinating solvents.
Two routes have been employed for the synthesis of Pd(II)/Pt(II) complexes of the type [MX′(PP2PO)]X′′ : a) The reaction of the trigonal-bipyramidal d8 systems [MX(PP3)]X6a,34–36 [M = Pd: X = Cl(1), M = Pt: X = Cl, Br (4, 5)] with H2O2 in chloroform (X′ = X′′= X: 7, 10, 11 shown in Scheme 1) and b) The reaction of [MX(PP3)]X [M = Pd: X = Cl, I (1, 3); M = Pt: X = Cl, I (4, 6)] in dimethylsulfoxide with aqueous GSH under acid conditions (X′ = X and M = Pd (7a, 7b); X′ = GS and M = Pd or Pt (7a′, 7b′ or 10a′, 10b′) shown in Scheme 2). The less common compounds [PdBr2(PP(PO)2)] (8) and [PtI2(PP(PO)2)] (12) were prepared by the reaction of [MX(PP3)]X [M = Pd: X = Br(2); M = Pt: X = I (6)] with H2O2 in chloroform (Scheme 1).
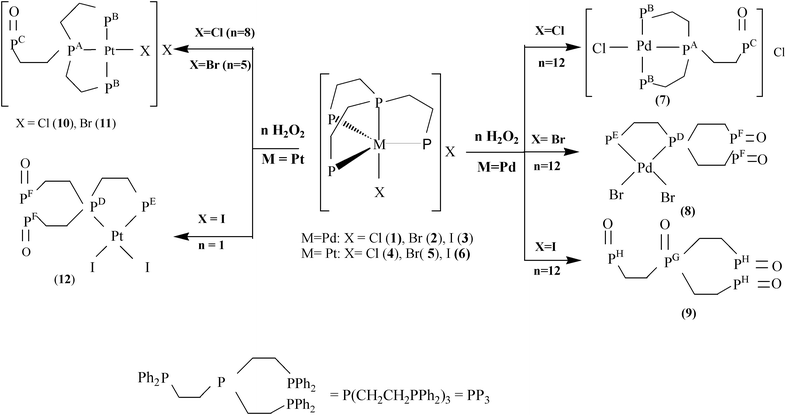 |
| | Scheme 1
Oxidations with H2O2 of complexes 1–6 in CDCl3. | |
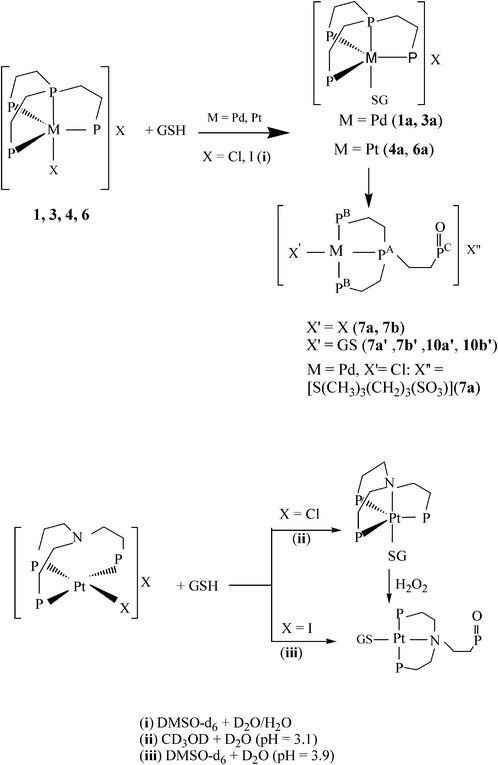 |
| | Scheme 2 Reactions with GSH for halo complexes containing the tripodal ligands PP3 and NP3. | |
The preparation of mononuclear complexes [Pd(PP(PO)2)2]X2 [X = Cl(13), Br (14)] exhibiting four dangling PO groups by reaction of 1/2 with PP333 and subsequent oxidation of [Pd(PP3)2]X2 [X = Cl(13*)/Br(14*)] with H2O2 (Scheme 3, eqn (1)) supported a dissociative mechanism for explaining the formation of PO groups rather than oxidations taking place at the coordinated P atoms.14a,37
| |  | (1) |
The entering PP
3 was able to originate the dissociation of two bound phosphino groups of
1/
2 leading to square-planar cationic complexes for which the X-ray crystal structure (Fig. S1, ESI
†) shows a square-planar PdP
4 arrangement, two
phosphines acting as bidentate chelating
ligands and four dangling arms available for
oxidation. The
mass spectra of
13/
14 confirm the 1
:
2 stoichiometry observing peaks attributable to [Pd(PP(PO)
2)
2]X and Pd(PP(PO)
2)
2 fragments. The
31P{
1H}NMR data in
DMSO-d6 (
Fig. 1) consist of three resonances exhibiting the expected 1(P
I)
:
1(P
J)
:
2 (P
K) integration ratio.
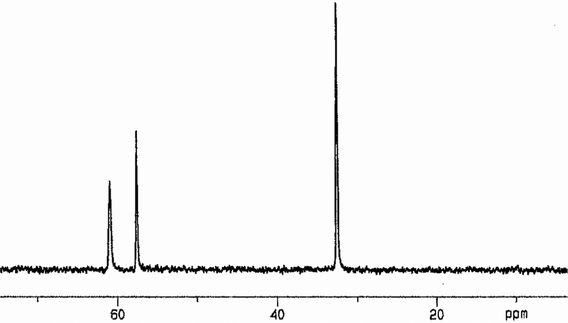 |
| | Fig. 1
31P{1H}NMR spectrum (r.t.) in DMSO-d6 for 13·3CH2Cl2. | |
Another example of dissociation of coordinated phosphorus in Pd(II) complexes containing the linear ligand triphos, that can occur with or without addition of phosphine depending on the halogen, is shown in Scheme 3. While it was necessary to add 1 equivalent of triphos to [PdCl(triphos)]Cl prior to affording the cationic species with a 1:2 Pd to triphos-O ratio, [PdBr(triphos)]Br reacted with H2O2 in acetone–ether (2:1) forming an ionic compound, where the cation contains two triphos-O ligands with a square-planar P4 donor set to Pd(II) and [PdBr4]2− is acting as counter anion (eqn S1, ESI†).
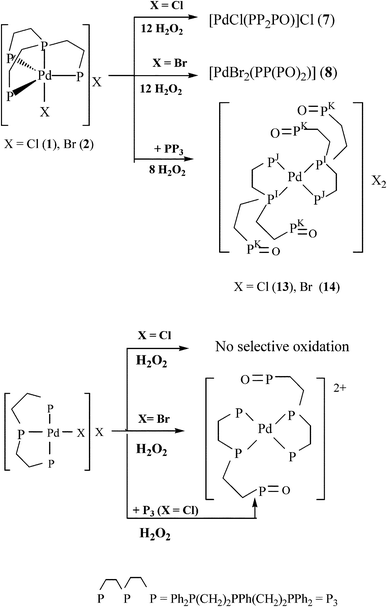 |
| | Scheme 3
Oxidations with H2O2 of palladium(II) complexes containing PP3 and the linear ligand triphos. | |
The related square-pyramidal complex [PdBr(triphos-O)2]Br, afforded from solutions of [PdBr(triphos)]Br in the same solvent system and characterized by X-ray diffraction,14e would be considered as an intermediate of the reaction where the weakly coordinated axial bromo ligand left the five-coordinate structure, giving the corresponding square-planar cation and counter anion. The X-ray analysis (Fig. 2, Tables 1 and 2) for single crystals of 13·6H2O·2CH3OH, obtained from DMSO solutions containing the solid of the reaction in CH3OH between 13·3CH2Cl2 and SnCl2, confirmed the structure predicted in solution. Inspection of the P–O bond distances (1.485(6) Å) reveals similar values to those found for [PdBr(triphos-O)2]Br (1.477(9)–1.457(10) Å)14e or [Pd(dppfcO)2]Cl2 (dppfcO = Ph2PC5H4P(O)Ph2) (1.487(3) Å)38 and lengthened distances with respect to the neutral square-planar compound [Pd(tripod–O0.5)Cl2] (tripod = CH3C(CH2PPh2)3; 1.39(2) Å).39 On this basis, the dissociation processes of coordinated PP3 responsible for the preparation of complexes 7–12 presumably occurred via a preassociative mechanism leading to equilibria (Scheme S1, ESI†) involving five- and four-coordinate species prior to oxidation of the resulting dangling phosphorus. The rapid exchange between chelate ring-opened and ring-closed forms seems to assure the appearance of pendant arms from both Pd(II) and Pt(II) precursors. However, the higher inertness of the dissociated species formed for M = Pt compared to Pd may explain the lower concentrations required of precursors 4–6 (ca. 3 × 10−3 M) compared to 1–3 (ca. 8 × 10−3 M). The 31P{1H}NMR spectrum of 7 (Fig. S2, ESI†) along with 10 (Fig. 3) and 11 consisted of three resonances showing integration ratios 1(PA): 2 (PB): 1 (PCO) in agreement with PP3 acting as tridentate chelating ligand and oxidation of just a terminal coordinated phosphorus of 1, 4 and 5, respectively (eqn (2)).
| |  | (2) |
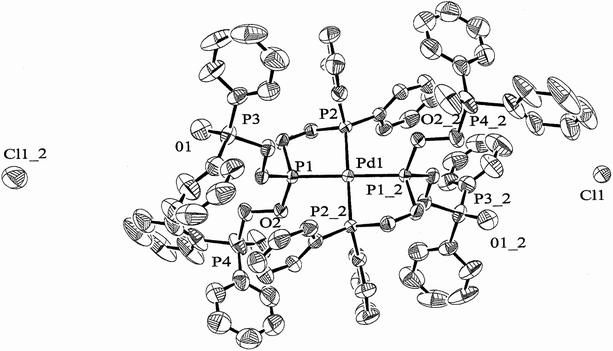 |
| | Fig. 2 ORTEP diagram for 13. 50% probability of thermal ellipsoid drawn and solvents removed for clarity. | |
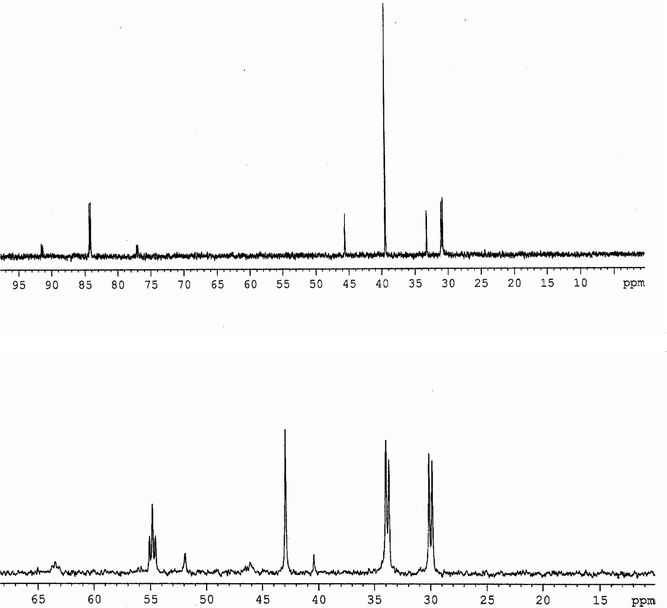 |
| | Fig. 3
31P{1H}NMR spectra (r.t.) in CDCl3 for 10·2H2O (top) and 10a·CHCl3 (bottom). | |
The coupling constants between PA and PC of 35(7), 39(10) and 37(11) Hz were as expected for distorted square-planar ionic complexes formed via a chelate ring-opening from the precursors.34–36 The 1J(31PA, 195Pt) and 1J(31PB, 195Pt) couplings for 10–11ca. 2900 and 2500 Hz are typical of PAtrans to X and PBtrans to each other, respectively, and consistent with 195Pt{1H}NMR data for 10 (Fig. S3, ESI†). Considering the enhancement of the dissociative character and steric hindrance going down the halogen group, and both the reductor behaviour and π-accepting ability of halide anions and ligands following the order I−> Br−> Cl−, one can explain, by the steric and electronic factors, why the extent of oxidations increases from Cl− or Br− to I−. Indeed, the 31P{1H}NMR spectra for 8 (Fig. S2, ESI†) and 12 (Fig. 4) exhibit three signals with an integration ratio 1(PD):1(PE):2(PFO) and couplings between PD and PF of 54 and 50 Hz, indicative of the formation of neutral species (eqn (3) and (4)) where the ligand is coordinated to the metal through the central and one terminal phosphorus and the two remaining dangling phosphorus are oxidized.
| |  | (3) |
| |  | (4) |
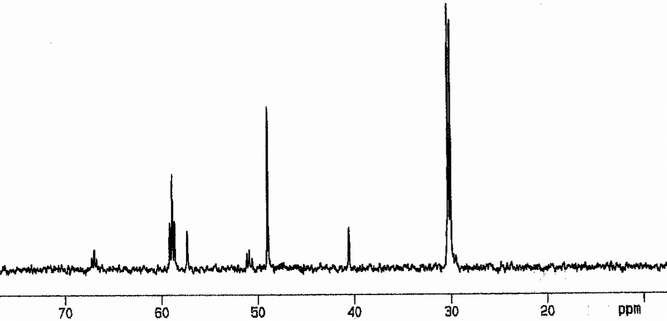 |
| | Fig. 4
31P{1H}NMR spectrum (r.t.) in CDCl3 for 12·H2O. | |
Concentrations of H2O2 greater than that used for the oxidation of 1 with 12 equivalents of H2O2 resulted in the appearance of three signals assigned to the ionic complex 7, as well as three resonances at δ 83.2, 65.1 and 31.2 ascribed to the neutral system [PdCl2(PP(PO)2)] bearing the same structure as 8 and 12. However, oxidation of complex 3, under similar conditions to 1 or 2, led to the appearance of two signals (Fig. S2, ESI†) with integration ratios 1(PG):3(PH) due to OP(PO)3 (9) (eqn (5)).
| | 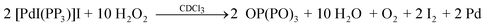 | (5) |
In consequence, complexes
7,
10–
11 seem to be the result of the action of H
2O
2 on the square-pyramidal intermediates I
a (having just one terminal pendant arm, Scheme S1, ESI
†), while the formation of
12 from
6 required the oxidation of I
a (by using the smallest concentration of H
2O
2) and subsequent evolution to I
b. Since in absence of H
2O
2 the
iodo complex
6 was stable in non deoxygenated chloroform, a mechanism whereby O
2 oxidized a terminal coordinated phosphorus of I
b was considered the most likely. The fact that the oxidation of
3 with less than 12 equivalents of H
2O
2 gave mixtures containing
3 and mono- and/or dioxidized complexes indicates rapid equilibria between dissociated and non dissociated forms that persisted by exposure to O
2 and disappeared to give
9 by increasing the amount of
hydrogen peroxide.
Although greater concentrations of H2O2 did not produce neutral species, with the same X2PtP2 arrangement to 12 (X = I), starting from the chlorocomplex 10 or the related compounds [MAuCl2(PP3)]Cl (M = Pd, Pt),34 the addition of [AuCl(tdg)] induced a chelate ring-opening affording [PtAuCl3(PP2PO)] (10a) with a chiral central phosphorus (Fig. 3) or [MAu2Cl4(PP3)] (Scheme 4). The increase in concentration of chloride ions lead to an associative mechanism via a trigonal-bipyramidal 18-electron intermediate (Ib) proposed for reaction mechanisms of common four-coordinate square-planar Pd(II) and Pt(II) compounds.40,41 However, the propensity of M′Cl salts (M′ = Cu, Ag) to undergo oligomerization with phosphines35 can justify the absence of ring-opening reactions by adding to 10 an excess of these chlorides (Fig. S4, ESI†). None of the reactions explored between [MX(PP3)]X in DMSO-d6 and GSH in D2O afforded neutral dioxidized complexes6c or the complete oxidation of PP3. The monoxidized systems coexisted in a mixture containing the unreacted (1) or regenerated (3, 6; vide infra) precursors and the trigonal-bipyramidal Pd(II)/Pt(II) complexes showing glutathionate (GS−) as a monodentate thiolato ligand (1a, 3a or 4a and 6a: Scheme 2), with the formation of 4a (M = Pt) as the predominant species needing a greater amount of GSH (Fig. S5, ESI†) than 1a (M = Pd).
 |
| | Scheme 4 Reactions of chloro complexes containing dangling PO and PAuCl groups. | |
The 31P{1H}NMR data for 1 + 1 eq. GSH and the spectrum for 1 + 2 eq. GSH, shown in Fig. 5, confirm the coexistence of 1, 1a and the two oxidized complexes [PdCl(PP2PO)][S(CH3)3(CH2)3(SO3)] (7a) and [Pd(SG)(PP2PO)]Cl (7a′). Single crystals of 7a·4H2O studied by X-ray diffraction were obtained by the addition of water to the sample for the reaction 1 + 3 eq. GSH (Fig. S6, ESI†), where the intensity of the signals due to 7a (with low solubility in H2O) and 1 (consumed in the formation of 7a) decreased by comparison with the spectrum for 1 + 2 eq. GSH. The solid state structure (Fig. 6 and Tables 1 and 2) is consistent with the presence of cations showing a distorted square-planar geometry with a ClP3 arrangement to Pd(II) and one dangling PO group. The P–O and Pd–P bond distances are in the same range and slightly shorter, respectively, than for 13. The average of 2.286(3) Å for Pd–P bond lengths agrees with the low trans influence of Cl compared to P and the two fused and non fused five-membered chelate rings imparted by 7a and 13, respectively. The S(1) and S(2) atoms of the counter anion are located below the distorted square-planar P(1)P(2)Pd(1)Cl(1)P(3) arrangement at 9.837 and 10.580 Å, respectively from Pd(1). Taking account of the relatively strong π-donating ability of thiolates to dissociate neighbouring coordinated phosphino groups, it is possible that one terminal phosphorus atom of 1a was dissociated, in part, by GS− giving the square-planar thiolate with a dangling phosphorus, followed by the irreversible oxidation (7a′) in non deoxygenated DMSO-d6 + D2O. Dimethylsulfoxide would produce the oxidation of the dissociated phosphorus undergoing reduction to sulphide with further formation of a trimethyl sulfonium fragment. This agrees with the formation of 7a from 7a′, in a process where the excess of reduced glutathione in the solvent system used presumably favored the glutathionato ligand substitution with chloride, subsequent fragmentation and oxidation of the thiol and generation of the propane sulfonato moiety of the [S(CH3)3(CH2)3(SO3)]− counter anion, instead of acting as a bulky entering ligand able to produce the neutral dioxidized complex [Pd(GS)2(PP(PO)2].
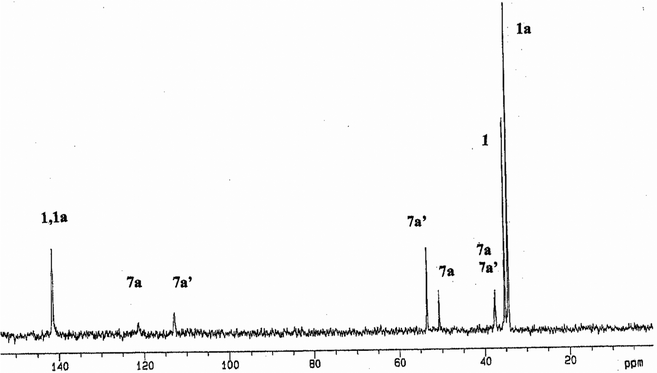 |
| | Fig. 5
31P{1H}NMR spectrum (r.t.) for 1 + 2 eq. GSH in DMSO-d6 + D2O (pH = 4.2). | |
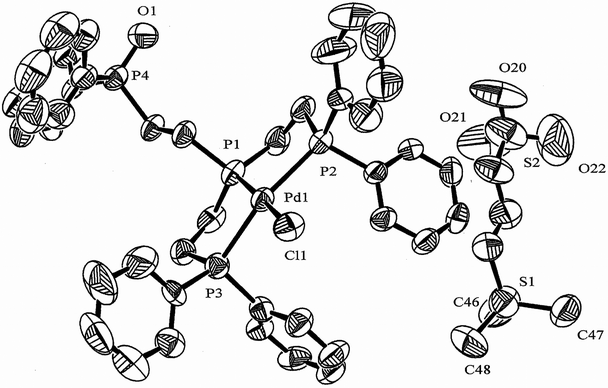 |
| | Fig. 6 ORTEP diagram for 7a. 50% probability of thermal ellipsoid drawn and solvents removed for clarity. | |
In the case of the reactions between 4 or 6 and GSH (followed by 31P (Experimental part) and 1H (Fig. S7 NMR, ESI†), the significant affinity of Pt(II) for the thiolato S atom prevents the appearance of square-planar species other than [Pt(GS)(PP2PO)]Cl(10a′) or [Pt(GS)(PP2PO)]I (10b′). In fact, the titration of 10 (in DMSO-d6) with aqueous GSH demonstrated the quantitative chloro ligand substitution reaction with glutathionato to give 10a′ from the first addition of thiol (Fig. S8, ESI†). The singlet at δ 98.5 assigned for both, 10a′ and 10b′, to the bound phosphorus PA, supports the absence of the square-planar halo complexes.
Table 1 Summary of crystal parameters, data collection, and refinement for 7a , 13∗ and 13
| Complex |
7a·4H2O |
13∗·4CHCl3 |
13·6H2O·2CH3OH |
| Empirical formula |
C48H57ClO8P4PdS2 |
C88H84Cl14P8Pd |
C86H90Cl2O12P8Pd
|
| Formula weight |
1091.79 |
1992.01 |
1740.64 |
|
T (K) |
293(2) |
293(2) |
293(2) |
| Wavelength (Å) |
0.71073 |
0.71073 |
0.71073 |
| Crystal size (mm) |
0.26 × 0.24 × 0.07 |
0.15 × 0.07 × 0.07 |
0.36 × 0.34 × 0.21 |
| Colour/habit |
Yellow/prisms |
Colorless/needles |
Colorless/prisms |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
C/2c |
P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
|
|
a (Å) |
36.211(5) |
14.942(2) |
11.808(6) |
|
b (Å) |
14.7523(19) |
18.073 (3) |
13.975(7) |
|
c (Å) |
20.890(3) |
17.298(3) |
15.675(7) |
|
α (°) |
90 |
90 |
105.495(7) |
|
β (°) |
106.565(3) |
93.440(4) |
98.909(7) |
|
γ (°) |
90 |
90 |
102.948(7) |
| Volume (Å3) |
10696(2) |
4662.8(13) |
2364.9 (19) |
| Z |
8 |
2 |
1 |
| Calculated density (Mg m−3) |
1.356 |
1.419 |
1.222 |
| Absorption coefficient (mm−1) |
0.642 |
0.780 |
0.441 |
|
F(000) |
4512 |
2032 |
902 |
|
θ range for data collection (°) |
1.17 to 24.71 |
1.63 to 26.41 |
1.38 to 26.43 |
| Index ranges |
−42 ≤ h ≤ 40 |
−18 ≤ h ≤ 18 |
−14 ≤ h ≤ 14 |
| 0 ≤ k ≤ 17 |
0 ≤ k ≤ 22 |
−17 ≤ k ≤ 16 |
| 0 ≤ l ≤ 24 |
0 ≤ l ≤ 21 |
0 ≤ l ≤ 19 |
| Reflections collected |
33386 |
28588 |
25012 |
| Independent reflections |
9091 [Rint = 0.0930] |
9528 [Rint = 0.0848] |
9623 [Rint = 0.0368] |
| Max. and min. transmission |
0.9564 and 0.8507 |
0.9474 and 0.8919 |
0.9131 and 0.8574 |
| Data/restraints/parameters |
9091/0/572 |
3947/0/502 |
7033/0/489 |
| Goodness of fit on F2 |
0.891 |
0.908 |
1.095 |
| Final R indices [I > 2σ(I)] |
R
1 = 0.0645 |
R
1 = 0.0672 |
R
1 = 0.0685 |
|
wR2 = 0.1523 |
0.1506 |
wR2 = 0.2029 |
| Largest diff. peak and hole (e Å−3) |
0.596 and −0.857 |
1.325 and −2.433 |
1.686 and −1.268 |
Table 2 Selected Distances (Å) and Angles (deg.) for 7a, 13∗ and 13
| Complex |
7a·4H2Oa |
13∗·4CHCl3a |
13·6H2O· 2CH3OHa |
|
Pd(1)⋯S(1) = 9.837 Å, Pd(1)⋯S(2) = 10.580 Å (7a),Pd(1)⋯Cl(1) = 12.640 Å (13∗), 15.357 Å (13).
|
| Pd(1)–P(1) |
2.217(3) |
2.3366(18) |
2.3406 (13) |
| Pd(1)–P(2) |
2.323(3) |
2.3231(19) |
2.3386(14) |
| Pd(1)–P(3) |
2.318(3) |
|
|
| Pd(1)–Cl(1) |
2.358(2) |
|
|
| P(3)–O(1) |
|
|
1.485(6) |
| P(4)–O(1) |
1.497(7) |
|
|
| P(4)–O(2) |
|
|
1.484(6) |
| S(2)–O(20) |
1.359(10) |
|
|
| S(2)–O(21) |
1.385(11) |
|
|
| S(2)–O(22) |
1.520(15) |
|
|
| S(1)–C(46) |
1.846(11) |
|
|
| S(1)–C(47) |
1.898(11) |
|
|
| S(1)–C(48) |
1.909(12) |
|
|
| P(1)–Pd(1)–P(2) |
84.23(10) |
83.62(7) |
83.67(5) |
| P(1)–Pd(1)–P(3) |
84.29(10) |
|
|
| P(2)–Pd(1)–P(3) |
157.28(9) |
|
|
| P(1)–Pd(1)–P(1_2) |
|
180.00(7) |
180.00(5) |
|
P(1).Pd(1)–P(2_2) |
|
96.38(7) |
96.33(5) |
| P(1)–Pd(1)–Cl(1) |
174.15(9) |
|
|
|
P(2)–Pd(1)Cl(1) |
97.51(9) |
|
|
| P(3)–Pd(1)–Cl(1) |
95 .89(9) |
|
|
The decisive role of dimethylsulfoxide in the oxidation processes leading not only to 7a, but also 7a′ and 10a′, was in accordance with the preparation of [M(SG)(PP3)]Cl·nH2O [n = 5 (M = Pd, 1a), 4 (M = Pt, 4a)] as solids (without signs of oxidized species in CD3OD)42 by interaction between solutions of 1 or 4 in methanol and aqueous GSH. Likewise, the coexistence in DMSO-d6 of [PdI(PP2PO)]I (7b) with 3 is in keeping with a behaviour as the oxidant of the solvent. In the presence of GSH, complexes 7b′ and 10b′ were formed via a five-coordinate intermediate Ia (Scheme S2, ESI†) from 3a and 6a, respectively. With excess GSH, both GS− and I− compete for the axial coordination site with the apparent prevalence of I− regenerating the cations of 3 and 6 (Fig. S9, ESI,† shows the prevalence of the resonance due to 3 as the concentrations of aqueous GSH were increased), in contrast with the behaviour observed for 1 and 4 (vide supra). However, the flexible coordination behaviour of the hemilabile ligand NP3 (Scheme S3, ESI†),43,44 seems to be responsible for the higher selectivity of both [PdCl(NP3)]Cl (which disappears by titration with GSH, Figure S10, ESI†) and [PtI(NP3)]I (Scheme 2) towards the square-planar thiolates containing one phosphine oxide group than the analogous PP3 compounds, 1 and 6.
Conclusions
The extent of the oxidations of [MX(PP3)]X with H2O2 in chloroform to afford ionic and neutral square-planar compounds increases from Cl− or Br− to I−, while in the presence of GSH and dimethylsulfoxide all oxidations conclude with the formation of ionic complexes containing just one PO arm. For M = Pt, the reaction with GSH led to [Pt(GS) (PP2PO)]X (X = Cl, I) and with H2O2 to [PtX(PP2PO)]X (X = Cl, Br) and [PtI2(PP(PO)2)]. Subsequent addition of [AuCl(tdg)], when X = Cl, produces the neutral species [PtAuCl3(PP2PO)]. For a metal as labile as Pd(II), [PdCl(PP3)]Cl gives a mixture of [Pd(GS)(PP2PO)]Cl + [PdCl(PP2PO)][S(CH3)3(CH2)3(SO3)] under reaction with GSH and [PdX(PP3)]X (X = Cl/Br) afford [PdCl(PP2PO)]Cl/[PdBr2(PP(PO)2] with H2O2, while the iodide forms [Pd(GS)(PP2PO)]I by adding GSH and OP(PO)3 by treatment with H2O2.
Experimental section
General procedures and instrumentation
Palladium chloride, palladium bromide and potassium tetrachloroplatinate were purchased from Strem Chemicals, copper chloride, silver chloride, silver nitrate, tris[2-(diphenylphosphino)ethyl]phosphine, 2,2′-thiodiethanol (tdg = thiodiglycol) and reduced form of glutathione from Aldrich, sodium chloride, bromide and iodide and H2O2 (30% w/v) from Panreac and gold metal from S.E.M.P.S.A. Microanalyses were performed on a Fisons Instrument EA 1108 CHNS–O. Liquid Secondary-Ion (LSI MS) mass spectra were obtained in a Micromass Autospec spectrometer using nitrobenzylic alcohol as the matrix. Infrared spectra were recorded at ambient temperature as KBr pellets (4000–500 cm−1) and Nujol mulls (500–100 cm−1) on a Mattson Cygnus 100 spectrophotometer. The bands are reported as vs = very strong, s = strong, m = medium, w = weak, sh = shoulder, br = broad. 1H, 31P{1H} and 195Pt {1H}NMR spectra were recorded on a Brucker AMX500 spectrometer at 500.1, 202.46 and 107.52 MHz, respectively. A 2 s relaxation delay and a 30 degree pulse angle were used to favor measurable integration data. Chemical shifts (δ) are reported in ppm relative to external sodium 3-(trimethylsilyl)propionate (1H), 85% H3PO4 (31P) and 1 M Na2PtCl6 (195Pt) ; s = singlet, d = doublet, t = triplet, q = quartet, m= multiplet, dd = doublet of doublets, dt = doublet of triplets, br = broad signal, J = coupling constant in Hz. Conductivities were measured at 25 °C using 10−3 M solutions in DMF on a WTW model LF-3 instrument. The pH values of the solutions were determined using a Corning 240 pH meter. The crystallizing solvents in the solids were confirmed by infrared and X-ray diffraction.
Solutions of Au(I)
Solutions containing [AuCl(tdg)] were prepared by procedures similar to those reported.45
Preparation of [MX(PP3)]X (1–6)
The precursors, 1–6, were prepared following the procedures previously described.34–36
Preparation of [MCl(PP2PO)]Cl [M = Pd (7), Pt (10)] and [PtBr(PP2PO)]Br (11)
Solutions containing [PdCl(PP3)]Cl (1) (red color, 0.0913 g, 0.1077 mmol), [PtCl(PP3)]Cl (4) (yellow color, 0.0894 g, 0.0954 mmol) and [PtBr(PP3)]Br (5) (yellow color, 0.0737, 0.07186 mmol) in CHCl3 [15 mL (1)/25 mL (4, 5)] were reacted with 1.2924, 0.7632 and 0.3397 mmol of H2O2, respectively. The mixtures of reaction were stirred at room temperature for 12, 18 and 2 h with formation of a suspension (7, 10) or a colorless solution (11). After that, Et2O (80 mL) was added to complete or provoke the precipitation. The solids were filtered off, washed with ether and dried in vacuo. 7·CHCl3 : Yield: 89%, white solid, mp 140–145 °C. Found: C, 52.8; H, 4.7. C43H43P4PdCl5O requires: C, 52.5; H, 4.4%. MS (LSIMS): m/z (rel. intensity) 862 (1%, M+), 827 (93%, M+ − Cl), 792 (5%, M+ − 2Cl), 579 (5%, M+ − 2Cl − (CH2)2PPh2). IRνmax/cm−1: (Pd–Cl) 310w, (Pd–P) 355w, 360w, (PdO) 1178 m. 31P{1H}NMR (298 K, DMSO-d6): δ 111.0d [1PA], 43.4s [2PB], 31.2d [1PCO]; 3J(PA–PC) = 35.0 Hz. Λ(DMF) = 66.3 ohm−1 cm2 mol−1. 10·2H2O: Yield: 88%, white solid, mp 195–205 °C. Found: C, 50.8; H, 4.8. C42H46P4PtCl2O3 requires: C, 51.0; H, 4.7%. MS (LSIMS): m/z (rel. intensity) 916 (91%, M+ − Cl), 881 (2%, M+ − 2Cl), 668 (3%, M+ − 2Cl − (CH2)2PPh2). IRνmax/cm−1: (Pt–Cl) 315 s, (Pt–P) 356w, (PO) 1182 m. 31P{1H}NMR (298 K, DMSO-d6): δ 84.2d [1PA], 39.4s [2PB], 30.8d [1PCO]; 3J(31PA–31PC) = 39 Hz; 1J(31PA–195Pt) = 2902 Hz; 1J(31PB–195Pt) = 2505 Hz. 195Pt NMR (298 K, DMSO-d6): δ 4829.6 dt; 1J(31PA–195Pt) = 2912 Hz; 1J(31PB–195Pt) = 2524 Hz. 11·2H2O : Yield 80%, white solid, mp 190–200 °C. Found: C, 45.4; H, 4.7. C42H46P4PtBr2O3 requires: C, 45.3; H, 4.5%. MS (LSIMS): m/z (rel. intensity) 960 (66%, M+ − Br), 881 (2%, M+ − 2Br), 668 (4% , M+ − 2Br − (CH2)2PPh2). IRνmax/cm−1:(Pt–Br) 254w, (Pt–P) 342 m, (PO) 1183 m. 31P{1H}NMR (298 K, DMSO-d6): δ 88.0 s [1PA], 39.9 s [2PB], 30.6d [1PCO]; 3J(31PA–31PC) = 37 Hz; 1J(31PA–195Pt) = 2900 Hz; 1J(31PB–195Pt) = 2474 Hz.
Preparation of [MX2(PP(PO)2)] [M = Pd: X = Br(8). M = Pt: X = I(12)]
Solutions containing [PdBr(PP3)]Br (2) (red, 0.1165 g, 0.1243 mmol) and [PtI(PP3)]I (6) (yellow, 0.1220 g, 0.1090 mmol) in CHCl3 [15 mL (2)/30 mL (6)] were treated with 1.4916 and 0.1090 mmol of H2O2, respectively. The reaction mixtures were stirred for 2 and 14 h, affording a yellow solution (8) and a suspension (12). Et2O (70 and 120 mL) was added to provoke and complete the precipitation. The solids were filtered off, washed with ether and dried in vacuo. 8·3H2O: Yield: 80%, yellow solid, mp 70–80 °C. Found: C, 49.0; H, 5.0. C42H48P4PdBr2O5 requires: C, 49,3; H, 4.7%. MS (LSIMS): m/z (rel. intensity) 950 (1%, M+), 871 (2%, M+ − Br), 579 (15% M+ − 2Br − (CH2)2P(O)Ph2). IRνmax/cm−1:(Pd–Br) 255 m, 250 sh, (Pd–P) 357 s, (PO) 1158vs. 31P{1H}NMR (298 K, DMSO-d6): δ 82.0t [1PD], 67.0d [1PE], 36.1d [2PFO]; 3J(31PD, 31PF) = 54 Hz. 12·H2O: Yield: 40%, beige solid, mp 170–180 °C. Found: C, 42.8; H, 3.6. C42H44P4PdI2O3 requires: C, 43.1; H, 3.8%. MS (LSIMS): m/z (rel. intensity) 1151 (5%, M+), 1024 (100%, M+ − I), 879 (3%, M+ − 2I), 668 (9%, M+ − 2I − (CH2)2P(O)Ph2). IRνmax/cm−1: (Pt–I) 155 s, 170 s, (PO) 1182 s. 31P{1H}NMR (298 K, DMSO-d6): δ 58.9t [1PD], 48.9s [1PE], 30.1d [2PFO]; 3J(31PD, 31PF) = 50 Hz; 1J(31PD–195Pt) = 3257 Hz; 1J(31PE–195Pt) = 3406 Hz.
Preparation of OP(PO)3 (9)
To a solution of PP3 (0.1890 g, 0.2810 mmol) in CH2Cl2 (10 mL), H2O2 (1.9412 mmol) was added. The reaction mixture was stirred for 3 h with formation of a precipitate by addition of Et2O (70 mL). The solid was filtered off, washed with ether and dried in vacuo. This oxide was also prepared by reaction of 3 (0.1300 mmol) in CDCl3 (15 mL) with 1.5600 mmol of H2O2. 9·3H2O: Yield: 92%, white solid, mp 190–200 °C. Found: C, 64.3; H, 6.0. C42H48P4O7 requires: C, 64.0; H, 6.1%. 31P{1H}NMR (298 K, DMSO-d6): δ 49.5q [1PGO], 32.0d [3 PHO]; 3J(31PG–31PH) = 48 Hz.
Titrations of 10 with CuCl, AgX (X = Cl, NO3) and [AuCl(tdg)]
To solutions of 10·2H2O in CDCl3, CuCl and AgCl as solids or solutions of AgNO3/[AuCl(tdg)] in CD3OD/CH3OH were added in different stoichiometric ratios. The reaction mixtures were stirred for 24 h and those involving AgX salts protected from the light. The AgCl precipitate formed (for titrations with AgNO3) was removed by filtration. The 31P{1H}NMR spectra of the solutions were recorded after each addition. 31P{1H}NMR (298 K, CDCl3 + CH3OH) : 10 + 1 eq. [AuCl(tdg)] δ 54.8t [1PM], 42.9 s [1PL], 33.8d [1PN = O], 30.0d [1PP]; 3J(31PM–31PN) = 51 Hz; 3J(31PM–31PP) = 56 Hz; 1J(31PM–195Pt) = 3522 Hz; 1J(31PL–195Pt) = 3637 Hz. (298 K, CDCl3 + CD3OD) : 10 + 1 eq. AgNO3δ 75.3d [1PA], 44.7s [2PB], 32.5d [1PCO]; 1J(31PA–195Pt) = 3218 Hz; 1J(31PB–195Pt) = 2574 Hz. 10 + 2 eq. AgNO3δ 76.2d [1PA], 45.0 s [2PB], 32.7d [1PCO]; 1J(31PA–195Pt) = 3269 Hz; 1J(31PB–195Pt) = 2571 Hz. (298 K, DMSO-d6): 10 + 1 eq. CuClδ 84.1d [1PA], 39.3 s [2PB], 31.1s [1PCO]; 1J(31PA–195Pt) = 2943 Hz; 1J(31PB–195Pt) = 2490 Hz. (298 K, CDCl3): 10 + 1 eq. AgClδ 84.2d [1PA], 39.4s [2PB], 30.7d [1PCO]; 1J(31PA–195Pt) = 2920 Hz; 1J(31PB–195Pt) = 2500 Hz.
Preparation of [PtAuCl3(PP2PO)] (10a)
A solution containing [PtCl(PP3)]Cl (4) (0.1239 g, 0.1320 mmol) in CHCl3 (30 mL) was treated with 0.9912 mmol of H2O2.The reaction mixture was stirred for 6 h. After that, a solution of [AuCl(tdg)] (0.0260 g of Au, 0.1320 mmol) in CH3OH (8 mL) was added dropwise. The resultant solution was stirred for 16 h. The solid formed, after addition of ether (80 mL), was removed by filtration and dried in vacuo. 10a·CHCl3: Yield: 65%, white solid, mp 100–107 °C. Found: C, 39.7; H, 3.3.C43H43P4PtCl6AuO requires: C, 39.6; H, 3.3%. MS (LSIMS): m/z (rel. intensity) 1148 (1%, M+ − Cl), 668 (3%, M+ − 3Cl − Au − (CH2)2PPh2). IRνmax/cm−1: (Pt–Cl) 303 m, (Au–Cl) 285 m, (Pt–P) 346w, 350w, (PO) 1169 m. 31P {1H}NMR (298 K, DMSO-d6): δ 55.0t [1PM], 42.5s [1PL], 33.9d [1PNO], 29.8d [1PP]; 3J(31PM–31PN) = 52 Hz; 3J(31PM–31PP) = 57 Hz; 1J(31PM–195Pt) = 3506 Hz; 1J(31PL–195Pt) = 3639 Hz
Preparation of [Pd(PP(PO)2)2]X2 [X = Cl (13), Br (14)]
To solutions of [PdCl(PP3)]Cl (1) (0.0586 g, 0.0692 mmol) and [PdBr(PP3)]Br (2) (0.0711 g, 0.0760 mmol) in CHCl3 (25 mL) solutions of PP3 [0.0464 g, 0.0692 mmol (X = Cl); 0.0509 g, 0.0760 mmol (X = Br)] in CHCl3 (20 mL) were added. The resultant mixtures were stirred for 12 h. The solids formed were filtered off, dissolved in 25 mL of CH2Cl2 and treated with H2O2 [0.5536 mmol (X = Cl), 0.6080 mmol (X = Br)]. The resultant suspensions were stirred for 16 h (X = Cl) and 8 h (X = Br). The final solids were filtered off, washed with ether and dried in vacuo. 13·3CH2Cl2: Yield: 73%, white solid, mp 140–150 °C. Found: C, 56.9; H, 4.9. C87H90P8PdCl8O4 requires: C, 56.7; H, 4.7%. MS (LSIMS): m/z (rel. intensity) 1545 (25%, M+ − Cl), 1510 (21%, M+ − 2Cl), 808 (18%, M+ − 2Cl − P(CH2CH2PPh2)(CH2CH2P(O)Ph2)2) 579 (21%, M+ − 2Cl − P(CH2CH2PPh2)(CH2CH2P(O)Ph2)2 − (CH2)2P(O)Ph2). IRνmax/cm−1: (Pd–P) 343 m, 356sh, (PO) 1179 m. 31P{1H}NMR (298 K, DMSO-d6): δ 61.0d [2PI], 57.6d [2PJ], 32.5dd [4PK=O]; 3J(31PI–31PJ) = 17 Hz; 3J(31PI–31PK) = 39 Hz. Λ(DMF) = 81.0 ohm−1 cm2 mol−1. 14·2H2O: Yield: 60%, white solid, mp 125–130 °C. Found: C, 59.0; H, 5.0. C84H88P8PdBr2O6 requires: C, 59.1; H, 5.2%. MS (LSIMS): m/z (rel. intensity) 1589 (25%, M+ − Br), 1510 (6%, M+ − 2Br), 808 (14%, M+ − 2Br − P(CH2CH2PPh2)(CH2CH2P(O)Ph2)2), 579 (22%, M+ − 2Br − P(CH2CH2PPh2)(CH2CH2P(O)Ph2)2 − (CH2)2P(O)Ph2). IRνmax/cm−1: (Pd–P) 349 m, 359 sh, (PO) 1184 s. 31P{1H}NMR (298 K, DMSO-d6): δ 59.2d [2 PI], 51.3d [2PJ], 30.1dd [4PKO]; 3J(31PI–31PJ) = 18 Hz; 3J(31PI–31PK) = 37 Hz. Λ (DMF) = 95.7 ohm−1 cm2 mol−1.
Titrations of 1, 3, 4 and 6 with reduced glutathione (GSH)
Solutions containing [PdX(PP3)]X [X = Cl (1), I (3)] and [PtX(PP3)]X [X = Cl (4), I (6)] in DMSO-d6 (0.6 mL) were treated with solutions containing one equivalent of GSH in D2O (0.3 mL). The products of reaction were stirred for 24 h and the precipitate (X = I) was removed by filtration. The pH was measured and the 31P and 1H (Figure S7, ESI†) NMR spectra were recorded. This procedure was repeated after additions of two and three molar equivalents of GSH in D2O (0.1 mL). 31P{1H}NMR (298 K, DMSO-d6 + D2O): (1 + 1 eq. GSH) (pH = 3.7) δ 141.6s [1Pax, 1 and 1a], 35.2s [3Peq ,1], 34.4s [3Peq, 1a], 122.0s [1PA, 7a], 50.6 s [2PB, 7a], 38.0d [1PCO, 7a and 7a′], 112.9s [1PA, 7a′], 53.4s [2PB, 7a′]; 3J(31PA–31PC) = 39 Hz. (3 + 1 eq. GSH) (pH = 4.9) δ 156.0 s [1Pax, 3 and 3a], 36.3 s [3Peq, 3], 34.3 s [3 Peq, 3a], 124.9 s [1PA, 7b], 52.0 s [2PB, 7b], 37.5 s [1 PCO, 7b and 7b′], 112.9 s [1 PA, 7b′], 53.4 s [2 PB, 7b′]. (4 + 3 eq. GSH) (pH = 4.2) δ 123.7 s [1Pax, 4], 30.9 s [3Peq, 4], 130.5 s [1Pax, 4a], 24.0 s [3Peq, 4a], 98.5d [1 PA, 10a′], 49.5 s [2PB, 10a′], 38.2d [1PCO, 10a′]; 3J(31PA–31PC) = 38 Hz (10a′); 1J(31Peq–195Pt) = 2570 Hz (4) 2616 Hz (4a); 1J(31Pax–195Pt) = 2007 Hz (4a); 1J(31PB–195Pt) = 2573 Hz (10a′). 6 + 2 eq. GSH) (pH = 5.4) 136.0br [1Pax, 6 and 6a], 23.7 s [3Peq, 6], 18.0s [3Peq, 6a], 98.5 s [1 PA, 10b′], 49.0 s [2 PB, 10b′], 38.0 s [1PCO, 10b′]; 1J(31Peq–195Pt) = 2574 Hz (6); 1J(31Pax–195Pt) = 2349 Hz (6a).
Preparation of crystals
Single crystals of 7a·4H2O were afforded by addition of H2O (0.3 mL) to a sample containing [PdCl(PP3)]Cl (0.0125 mmol) + 3 eq. GSH in DMSO-d6 + D2O (0.6 + 0.5 mL), collected by filtration, washed with ether and dried in vacuo. Crystals of 13∗·4CHCl3 were prepared by reaction of [PdAgCl3(PP3)]35 (0.0195 mmol in 2 mL CDCl3) with four equivalents of PP3 (1 mL CDCl3), filtered off, washed with ether and dried in vacuo. Crystals of 13·6H2O·2CH3OH, used for X-ray diffraction, were prepared by addition of 6 equivalents of SnCl2 to 13·3CH2Cl2 (0.0150 mmol) in 2 mL of CH3OH, removal of the white solid formed and recrystallization in DMSO (2 mL).
Yellow and colorless prisms of 7a·4H2O and 13·6H2O·2CH3OH, respectively, and colorless needles of 13∗·4CHCl3 were mounted on glass fibers and used for data collection. Crystal data were collected at 293(2) K using a BRUKER SMART CCD 1000 diffractometer. Graphite monochromated Mo-Kα radiation was used throughout. The quality of the 7a·4H2O crystals did not allow a larger theta-range The data were processed with SAINT46 and empirical absorption correction was made using SADABS.47 The structures were solved by direct methods using the SIR-92.48 The structures were refined by full-matrix least-squares techniques against F2 using SHELXL-97.49 Positional and anisotropic displacement parameters were refined for all non-hydrogen atoms. Hydrogen atoms were placed geometrically, and positional parameters were refined using a riding model. Atomic scattering factors were obtained with the use of International Tables for X-ray Crystallography.50 Molecular graphics were obtained from ORTEP-3 for windows.51
Acknowledgements
We thank Xunta de Galicia (Spain) for financial support.
References
- S. Dinda, M. G. B. Drew and R. Bhattacharyya, Catal. Commun., 2009, 10, 720 Search PubMed.
- L. Wu, Z.-W. Li, F. Zhang, Y.-M. He and Q.-H. Fan, Adv. Synth. Catal., 2008, 350, 846 CrossRef CAS.
-
(a) F. Caruso, C. Pettinari, F. Paduano, R. Villa, F. Marchetti, E. Monti and M. Rossi, J. Med. Chem., 2008, 51, 1584 CrossRef CAS;
(b) S. Aizawa, T. Sano and Y. Fujita, J. Organomet. Chem., 2008, 693, 3638 Search PubMed;
(c) R. Kizek, J. Vacek, L. Trnkova and F. Jelen, Bioelectrochemistry, 2004, 63, 19 CrossRef CAS.
- S.-M. Kuang, P. E. Fanwick and R. A. Walton, Inorg. Chim. Acta, 2002, 338, 219 CrossRef CAS.
- J. Li and T. S. A. Hor, Dalton Trans., 2008, 5708 RSC.
-
(a) D. Fernández, M. I. García-Seijo, T. Kégl, G. Petöcz, L. Kóllar and M. E. García-Fernández, Inorg. Chem., 2002, 41, 4435 CrossRef CAS;
(b) S. Aizawa, K. Saito, T. Kawamoto and E. Matsumoto, Inorg. Chem., 2006, 45, 4859 CrossRef CAS;
(c) S. Aizawa, T. Kawamoto and K. Saito, Inorg. Chim. Acta, 2004, 357, 2191 Search PubMed;
(d) S. Aizawa, Y. Sone, T. Kawamoto, S. Yamada and M. Nakamura, Inorg. Chim. Acta, 2002, 338, 235 CrossRef CAS;
(e) S. Aizawa, T. Kobayashi and T. Kawamoto, Inorg. Chim. Acta, 2005, 358, 2319 CrossRef CAS.
- A .F. M. Mustafizur Rahman, T. Murafuji, T. Shibasaki, K. Suetake, K. Kurotobi and Y. Sugihara, Organometallics, 2007, 26, 2971 Search PubMed.
-
(a) S. Kealey, N. J. Long, P. W. Miller, A. J. P. White, P. B. Hitchcock and A. Gee, Dalton Trans., 2007, 2823 RSC;
(b) E. Bonfada, E. Schulz- lang, R. A. Zan and U. Abram, Z. Naturforsch., 2000, 55b, 285 Search PubMed;
(c) W. O. Siegl, S. J. Lapporte and J. P. Collman, Inorg. Chem., 1971, 10, 2158 CrossRef CAS;
(d) P. A. W. Dean, D. D. Phillips and L. Polensek, Can. J. Chem., 1981, 59, 50 Search PubMed;
(e) C. A. Ghilardi, P. Innocenti, S. Midollini and A. Orlandini, J. Chem. Soc., Dalton Trans., 1986, 2076 Search PubMed;
(f) S. M. Aucott, A. M. Z. Slawin and J. D. Woollins, J. Chem. Soc., Dalton Trans., 2000, 2559 RSC.
- E. I. Tolis, K. A. Vallianatou, F. J. Andreadaki and D. I. Kostas, Appl. Organomet. Chem., 2006, 20, 335 Search PubMed.
- L. R. Pignotti, N. Kongrakaiwoot, W. W. Brennessel, J. Baltrusaitis and R. L. Luck, J. Organomet. Chem., 2008, 693, 3263 Search PubMed.
- Z. P. Pai, P. V. Berdnikova, A. G. Tolstikov, O. N. Roor, T. B. Khlebnikova, N. K Gusarova, S. F. Malysheva, N. I. Ivanova and B. A. Trofimov, React. Kinet. Catal. Lett., 2009, 94, 319 Search PubMed.
- A. S. King, G. Ferguson, J. F. Britten and J. F. Valliant, Inorg. Chem., 2004, 43, 3507 CrossRef CAS.
- S. Priya, M. S. Balakrishna, J. T. Mague and S. M. Mobin, Inorg. Chem., 2003, 42, 1272 CrossRef CAS.
-
(a) L.F. Szczepura, B.A. Ooro and S.R. Wilson, J. Chem. Soc., Dalton Trans., 2002, 3112 RSC;
(b) F. Cecconi, C. A. Ghilardi, S. Midollini, S. Moneti, A. Orlandini and G. Scapacci, J. Chem. Soc., Dalton Trans., 1989, 211 RSC;
(c) H. Heinze, G. Huttner and L. Zsolnai, Chem. Ber., 1997, 130, 1393 CrossRef CAS;
(d) C. A. Ghilardi and L. Sacconi, Cryst. Struct. Commun., 1975, 4, 687 CAS;
(e) P. Sevillano, A. Habtemariam, A. Castiñeiras, M. E. García and P. J. Sadler, Polyhedron, 1998, 18, 383 CrossRef.
- G. Du and J. H. Espenson, Inorg. Chem., 2005, 44, 2465 CrossRef CAS.
- A. Cervilla, F. Pérez-Pla, E. LLopis and M. Piles, Inorg. Chem., 2006, 45, 7357 Search PubMed.
- K. Motoshima, A. Sato, H. Yorimitsu and K. Oshima, Bull. Chem. Soc. Jpn., 2007, 80, 2229 Search PubMed.
- M. J. Ingleson, M. Pink, H. Fan and K. G. Caulton, Inorg. Chem., 2007, 46, 10321 CrossRef CAS.
- A. Sen and J. Halpern, J. Am. Chem. Soc., 1977, 99, 8337 CrossRef CAS.
- A. Dovletoglou, S. A. Adeyemi, M. H. Lynn, D. J. Hodgson and T. J. Meyer, J. Am. Chem. Soc., 1990, 112, 8989 CrossRef CAS.
- R. Huang and J. H. Espenson, Inorg. Chem., 2001, 40, 994 CrossRef CAS.
- T. Yamada, K. Suzuki, K. Hashimoto and T. Ikeno, Chem. Lett., 1999, 28, 1043 Search PubMed.
-
(a) C. Stoll, I. P. Lorenz, H. Nöth and W. Ponikwar, J. Organomet. Chem., 2000, 602, 24 CrossRef CAS;
(b) M. Makosza, M. Paszewski and D. Sulikows Ki, Synlett, 2008, 19, 2938 Search PubMed.
-
(a) G. Titvinidze, A. Kaltbeitzel, A. Manhart and W. H. Meyer, Fuel Cells, 2010, 10, 390 Search PubMed;
(b) S.-I. Kawaguchi, M. Kotani, T. Ohe, S. Nagata, A. Namoto, M. Sonoda and A. Ogawa, Phosphorus, Sulfur Silicon Relat. Elem., 2010, 185, 1090 Search PubMed;
(c) A. V. Firth, S. W. Haggata, P. K. Khanna, S. J. Williams, J. W. Allen, S. W. Magennis, I. D. W. Samuel and D. J. Cole-Hamilton, J. Lumin., 2004, 109, 163 Search PubMed;
(d) G. A. Kostin, T. V. Us, J. M. Korda, V. G. Torgov, N. V. Kuratieva, S. I. Miroshnichenko and V. I. K. Alchenko, J. Inclusion Phenom. Macrocyclic Chem., 2010, 68, 131 Search PubMed;
(e) R. C. J. Atkinson, V. C. Gibson, N. J. Long and A. J. P. White, Dalton Trans., 2010, 39, 7540 RSC;
(f) H.-S. Son and J.-Y. Lee, Org. Electron., 2011, 12, 1025 CrossRef CAS;
(g) W. Levason, M. E. Light, S. Maheshwari, G. Reid and W. Zhang, Dalton Trans., 2011, 40, 5291 RSC;
(h) C. J. Lomoschitz, B. Feichtenschlager, N. Moszner, M. Puchberger, K. Moller, M. Abele and G. Kickelbick, Langmuir, 2011, 27, 3534 Search PubMed;
(i) R. Ahmed, A. Altieri, D. M. D'Souza, D. A. Leigh, K. M. Mullen, M. Papmeyer, A. M. Z. Slawin, J. K. Y. Wong and J. D. Woolins, J. Am. Chem. Soc., 2011, 133, 12304 Search PubMed.
- M. Baaden, G. Wipff, M. R. Yaftian, M. Burgard and D. Matt, J. Chem. Soc., Perkin Trans. 2, 2000, 7, 1315 Search PubMed.
- M. R. Yaftian, M. Burgard, A. El Bachiri, D. Matt, C. Wieser and C. B. Dieleman, J. Inclusion Phenom. Mol. Recognit. Chem., 1997, 29, 137 CrossRef CAS.
- J. Yin, Y. Lin, X. Cao, G. A.Yu, H. Tu and S. H. Liu, Dyes Pigm., 2009, 81, 152 CrossRef CAS.
- K.-H. Kim, S. Jang and F. W. Harris, Macromolecules, 2001, 34, 8925 Search PubMed.
- E. Bergin, C. T. O'Connor, S. B. Robinson, E. M. McGarrigle, C. P. O'Mahony and D. G. Gilheany, J. Am. Chem. Soc., 2007, 129, 9566 CrossRef CAS.
- M. O. Shulyupin, I. G. Trostyanskaya, M. A. Kazankova and I. P. Beletskaya, Russ. J. Org. Chem., 2006, 42, 17 Search PubMed.
- M. Bertoth, C. Saluzzo, G. Mignani and M. Lemaire, Tetrahedron: Asymmetry, 2004, 15, 639 CrossRef CAS.
- P. Barbaro, C. Bianchini, G. Giambastiani and A. Togni, Chem. Commun., 2002, 2672 RSC.
- D. Fernández-Anca, M. I. García-Seijo and M. E. García-Fernández, Dalton Trans., 2010, 39, 2327 RSC.
- D. Fernández, M. I. García-Seijo, A. Castiñeiras and M. E. García-Fernández, Dalton Trans., 2004, 2526 RSC.
- D. Fernández-Anca, M. I. García-Seijo, A. Castiñeiras and M. E. García-Fernández, Inorg. Chem., 2008, 47, 5685 CrossRef CAS.
- D. Fernández, M. I. García-Seijo, P. Sevillano, A. Castiñeiras and M. E. García-Fernández, Inorg. Chim. Acta, 2005, 358, 2575 CrossRef CAS.
- Q-B. Bao, S. J. Landon, A. L. Rheingold, T. M. Haller and T. B. Brill, Inorg. Chem., 1985, 24, 900 CrossRef CAS.
- R. J. Coyle, Y. L. Slovokhotov, M. Y. Antipin and V. V. Grushin, Polyhedron, 1998, 17, 3059 CrossRef CAS.
- P. Sevillano, A. Habtemariam, M. I. García-Seijo, A. Castiñeiras, S. Parsons, M. E. García and P. J. Sadler, Aust. J. Chem., 2000, 53, 635 CAS.
-
R. G. Wilkins, Kinetics and Mechanisms of Reaction of Transition Metal Complexes, 2nd. Ed; VCH: Weinheim, Germany, 1991; Chapter 4 Search PubMed.
- D. Fernández, P. Sevillano, M. I. García-Seijo, A. Castiñeiras, L. Jánosi, Z. Berente, L. Kollár and M. E. García-Fernández, Inorg. Chim. Acta, 2001, 312, 40 CrossRef CAS.
-
31P{1H}NMR (CD3OD): (1a) δ 137.8 s (Pax), 33.0 s (Peq); (4a) δ 129.7 s (Pax), 22.6 s (Peq), 1J(31Pax–195Pt) = 2113 Hz; 1J(31Peq–195Pt) = 2617 Hz.
- M. I. García-Seijo, A. Habtemariam, P. del S. Murdoch, R. O. Gould and M. E. García-Fernández, Inorg. Chim. Acta, 2002, 335, 52 CrossRef CAS.
- M. I. García-Seijo, A. Habtemariam, D. Fernández-Anca, S. Parsons and M. E. García-Fernández, Z. Anorg. Allg. Chem., 2002, 628, 1075 CrossRef CAS.
- P. Sevillano, A. Habtemariam, S. Parsons, A. Castiñeiras, M. E. García and P. J. Sadler, J. Chem. Soc., Dalton Trans., 1999, 2861 RSC.
- SMART and SAINT, Area Detector Control and Integration Software; Bruker Analytical X-ray Instruments Inc.: Madison, WI, 1997 Search PubMed.
-
G. M. Sheldrick, SADABS, Program for Empirical Absorption Correction of Area Detector Data; University of Goettingen, Germany, 1997 Search PubMed.
- A. Altomare, G. L. Cascarano, C. Giacovazzo and A. Guagliardi, SIR-92, A program for crystal structure solution., J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
-
(a)
G. M. Sheldrick, SHELX-97. An integrated system for solving and refining crystal structures from diffraction data. University of Goettingen, Germany, 1997 Search PubMed;
(b) G. M. Sheldrik, A short history of SHELX. Acta Cryst, 2008, A64, 112 Search PubMed.
-
International Tables for X-ray Crystallography; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1995, Vol. C Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.