Tetrathiafulvalene as a one-electron iodine-free organic redox mediator in electrolytes for dye-sensitized solar cells†
Ze
Yu
a,
Haining
Tian
b,
Erik
Gabrielsson
b,
Gerrit
Boschloo
c,
Mikhail
Gorlov
b,
Licheng
Sun
b and
Lars
Kloo
*a
aInorganic Chemistry, Royal Institute of Technology, S-100 44, Stockholm, Sweden
bOrganic Chemistry, Royal Institute of Technology, S-100 44, Stockholm, Sweden
cPhysical Chemistry, Uppsala University, Box 259, S-751 05, Uppsala, Sweden
First published on 5th December 2011
Abstract
Tetrathiafulvalene (TTF) was investigated as an organic iodine-free redox mediator in electrolytes for dye-sensitized, nanocrystalline solar cells (DSCs) and was compared to the commonly used iodide/triiodide system. The TTF system studied was determined to be a one-electron transfer system, although potentially exhibiting three well-defined oxidation states. Despite the slightly positive redox potential of TTF, electrolytes with TTF displayed around 200 mV lower open-circuit voltage than the iodide/triiodide system. This can mainly be ascribed to a much shorter electron lifetime in the TiO2 film. Mass transport limitations for redox species in TTF-based electrolytes were found to be serious. Electrochemical impedance measurements (EIS) show that the charge-transfer resistance at the counter electrode in the electrolyte with TTF is considerably larger than for the iodide/triiodide system. In addition, the light absorption of the TTF-based electrolyte is stronger than that for the iodide/triiodide system. Thus, DSCs with TTF-based electrolytes show worse photovoltaic performance than those with iodide/triiodide-based electrolytes. The differences in I–V characteristics and charge-recombination behavior have also been elucidated.
Introduction
Dye-sensitized nanocrystalline solar cells (DSCs) were first reported in 1991,1 and during the past two decades they have attracted a great deal of interest because of their relatively low cost of production in contrast to silicon-based photovoltaic devices.2–4 The redox mediator (redox couple) dissolved in a liquid electrolyte solution handles the charge transport between the working and counter electrodes. The iodide/triiodide (I−/I3−) system has been used as a redox mediator from the very beginning of DSC research, and has turned out to be the most useful redox mediator so far. The highest photovoltaic conversion efficiency achieved is over 11% using I−/I3−.5,6 However, some properties of the I−/I3− system are not satisfactory; for instance, light absorption of I3− in the part of the visible light spectrum and large energy loss upon dye regeneration, which further limits improvement of the photovoltaic performance. In addition, there are two other main drawbacks of the I−/I3− system, the corrosion of many metals and the vapor pressure of iodine itself. These properties severely limit the practical application of DSCs in the view of long-term durability. Therefore, a large number of attempts have been made over the past years to replace the I−/I3− system with less absorbing, less corroding and more stable alternatives. Several organometallic complexes have been tested as alternative redox mediators for DSCs.2,4,7–12 Moreover, many systems, such as ferricenium/ferrocene (Fc/Fc+),13SCN−/(SCN)3−,14SeCN−/(SeCN)3−,14,15 the organic radical 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO)16 and others, have been used to replace the I−/I3− system. Their photovoltaic performance, however, could not match that of I−/I3−. The main reasons are largely due to the rapid electron recombination between photoinjected electrons in TiO2 and the oxidized form of the redox couple in the electrolyte and/or slower dye regeneration.17 Recently, several impressive improvements have been achieved by using organic sensitizers in conjunction with Fc/Fc+,18copper (I/II)19 and cobalt (II/III)20 complexes; the conversion efficiencies of which rival those for iodine-based electrolytes. Organic, two-electron thiolate/disulfide redox mediators have also been intensely investigated.21–23 In combination with ruthenium or organic sensitizers, comparable conversion efficiencies have been achieved by using such thiolate/disulfide redox mediators.21,23Tetrathiafulvalene (TTF) is an organosulfur compound and has been extensively investigated as electron-donating unit in charge-transfer complexes.24–28TTF may be oxidized in two steps to TTF2+, the redox potentials of which both fit the potential/energy range for a suitable redox mediator in a DSC. This makes TTF a suitable candidate as an alternative redox mediator for DSCs. Zhang et al. studied TTF as an organic iodine-free redox mediator in DSCs in conjunction with one ruthenium dye a few years ago, and some preliminary photovoltaic performance results were included into his doctoral thesis.29 We herein further investigate TTF in combination with a bulky organic dye, D35 (Fig. 1).30 For comparison, I−/I3−-based electrolytes were also included in this study, in which the corresponding concentrations of I− or I3− and the ratio between oxidized/reduced species were kept the same as in the TTF-based electrolytes. Our aim has been to compare the performance of the TTF and I−/I3− systems on the photovoltaic performance and, more importantly, to gain a deeper insight from such comparisons.
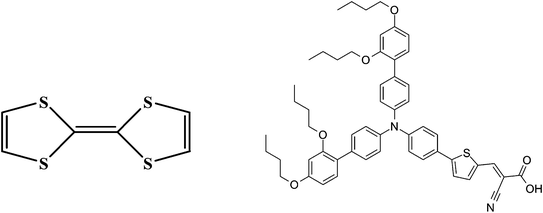 | ||
| Fig. 1 Structures of TTF (left) and the organic dye D35 (right). | ||
Experimental section
Reagents and electrolytes
Acetonitrile (AN), iodine, 1-propyl-3-methylimidazolium iodide (PMII), lithium trifluoromethanesulfonate (lithium triflate), nitrosonium tetrafluoroborate (NO[BF4]), tert-butyl pyridine (4-TBP) and ethanol were purchased from Aldrich. The synthesis of the dye D35 (E)-3-(5-(4-(Bis(20,40-dibutoxybiphenyl-4-yl)amino)phenyl)thiophen-2-yl)-2-cyanoacrylic acid has been published previously.30Dye N-719 cis-RuL2(SCN)2 (L = 2,2′-bipyridyl-4,4′-dicarboxylic acid) was purchased from Solaronix. The TTF-based electrolyte (ET) was made as follows: 0.2 M TTF, 0.02 M NO[BF4], 0.1 M lithium triflate and 0.5 M 4-TBP in AN; The composition of the iodine-based electrolyte EI was: 0.2 M PMII, 0.02 M I2, 0.1 M lithium triflate and 0.5 M 4-TBP in AN.Device fabrication
The fabrication of the solar cell devices has been described previously.31 A ∼2 μm thick film of 25 nm-sized TiO2 nanoparticle paste was first printed on the conducting glass substrates, followed by printing of a scattering layer (∼4 μm, PST-400C, JGC Catalysts and Chemicals LTD) to render a final thickness of ∼6 μm. The prepared working electrodes were heated at 300 °C for 10 min before use. After cooling to 80 °C, the working electrode was immersed into a 0.2 mM solution of D35 in ethanol solution at room temperature for at least 12 h. The sensitized working electrodes were assembled with a platinized counter electrode (CE) using a 25 μm hot-melt Surlyn frame. The electrolyte was introduced under vacuum through a prefabricated drilled hole in the CE. The hole was sealed afterwards with a 50 μm thick thermoplastic frame and a glass plate. DSCs were fabricated with the electrolytes ET and EI, respectively. Six solar cells were prepared, three of each electrolyte composition, showing highly reproducible results. The active area of the cells was 0.25 cmCurrent–voltage characteristics
A solar simulator (Newport 91160-1000) was used to give an irradiation of 100 mW cm−2 [the equivalent of 1 sun at air mass (AM) 1.5G]. The overall light-to-electricity conversion efficiency (η), fill factor (FF), open-circuit voltage (Voc), and short-circuit current density (Jsc) were obtained through the current–voltage characteristics of the solar cells. These characteristics and I–V diagrams were recorded using a computerized Keithley model 2400 source/meter unit. The photovoltaic performance was determined by using a plastic mask ∼1 mm wider than the active area. Three cells based on each electrolyte were investigated, showing high reproducibility. The I–V data listed in Table 1 are the average values of the two best cells based on each electrolyte studied. The incident photon-to-current efficiencies (IPCE) measurements were determined using a computerized set-up consisting of a xenon arc lamp (300 W Cermax, ILC Technology), followed by a 1/8 m monochromator (CVI Digikrom CM 110). The data were collected by a Keithley 2400 source/meter. Both the solar simulator and the IPCE system were calibrated using a certified solar cell (Fraunhofer ISE).| Electrolytes | Light intensitya (W m−2) | J sc (mA cm−2) | V oc (V) | FF | η (%) |
|---|---|---|---|---|---|
| a 1000 W m−2, 500 W m−2 and 100 W m−2 equal to 1 sun, 0.5 sun and 0.1 sun, respectively. | |||||
| ET | 1000 | 4.29 | 0.65 | 0.45 | 1.24 |
| 500 | 2.91 | 0.62 | 0.65 | 2.32 | |
| 100 | 0.64 | 0.55 | 0.50 | 1.73 | |
| EI | 1000 | 12.40 | 0.82 | 0.64 | 6.50 |
| 500 | 6.54 | 0.80 | 0.69 | 7.17 | |
| 100 | 1.14 | 0.75 | 0.75 | 6.39 |
Photoelectrochemical and electrochemical impedance measurements
Photoelectrochemical measurements were performed by using a green light-emitting diode (Luxeon K2 star 5W, λmax = 530 nm) as the light source. Voltage and current traces were recorded by a 16-bit resolution data acquisition board (National Instruments) in combination with a current amplifier (Stanford Research SR570). The relation between potential and charge was studied using a combined voltage decay/charge-extraction method. Electron lifetimes were studied by monitoring the transient photovoltage response after a small perturbation of light intensity.32 The electrochemical potential EF,n in TiO2 was derived from the equation Voc = |EF,n−Eredox|, where Voc is the open-circuit voltage of the DSC and Eredox for electrolytes ET and EI are 0.56 V and 0.35 V17vs.NHE, respectively. The data used in the figures for photoelectrochemical measurements were based on the cells that showed the best performance of each electrolyte. Impedance measurements were carried out using an Autolab PGstat12 potentiostat with an impedance module. The frequency range used was 10 kHz to 0.1 Hz, using 20 mV AC amplitude. Impedance was recorded for the electrolytes ET and EI in the dark with zero bias voltage.Electrochemical measurements
Electrochemical measurements were carried out by using an Autolab potentiostat with a GPES electrochemical interface (Eco Chemie) at a scan rate of 100 mV s−1. Cyclic voltammograms were obtained by dissolving 1 mM of TTF in AN together with 0.1 M tetrabutylammonium hexafluorophosphate (Fluka, electrochemical grade) as supporting electrolyte. The working electrode was a freshly polished glassy carbon disk, the reference electrode was a non-aqueous Ag/Ag+ electrode (0.01 M AgNO3 in AN), and a platinum wire was used as auxiliary electrode. The reference electrode was calibrated against the Fc/Fc+ couple and potentials were converted to the normal hydrogen electrode (NHE) scale by using a value of 630 mV vs.NHE for Fc+/Fc.33 Note that all the potentials mentioned in the following part are with respect to NHE.UV-vis measurements
The UV-vis absorption spectra of 100 × diluted electrolytes ET and EI were recorded on a Lambda 750 (PerkinElmer) UV-Vis spectrophotometer using a 1 cm pathlength quartz cuvette.Transient absorption spectroscopy
The ns-laser spectrometer used consists of a frequency tripled Nd:YAG laser (Continuum Surelight II, 10 Hz repetition rate, pulse width 10 ns) in combination with an OPO (Continuum Surelight), and an Edinburgh Instrument LP920 laser flash photolysis spectrometer, equipped an ICCD camera (Andor Technology) for spectral analysis. Laser pulses at 530 nm were attenuated to 1 mJ cm−2 per pulse by a system using a movable λ/2 plate and a fixed polarizer. The inert electrolyte consists of 0.1 M lithium triflate and 0.5 M 4-TBP in AN. In the case of the TTF-based electrolyte, 0.2 M TTF was added into the inert electrolyte.Results and discussion
The cyclic voltammogram of TTF in AN solution is presented in Fig. S1 in the ESI.† Two oxidation processes can be observed forming the radical cation TTF+ and the dication TTF2+ at 0.56 V and 0.91 V vs.NHE, respectively. By varying the concentrations of the oxidized form, potentiometric measurements were carried out to detect the number of transferring electrons involved in the system studied (Fig S2, ESI†). According to the Nernst equation, the number of electrons involved in the TTF→TTF+ oxidation corresponds to a quasi-one electron system. Since the second redox potential (0.9 V) is close to the HOMO-level of dye D35 (1.04 V)30, the driving force for dye regeneration is expected to be insufficient. Consequently, the TTF/TTF+ process is assumed to be the operating redox process for electrolyte ET in this work. This means that the TTF system still acts as a one-electron redox mediator, in contrast to the previously studied thiolate/disulfide systems.21–23 The actual redox potential of TTF/TTF+ and I−/I3− are 0.50 V and 0.37 V, respectively, according to the Nernst equation, where n is 1 for electrolyte ET and 2 for electrolyte EI.The TTF-based electrolyte ET was first used in DSCs in combination with the standard ruthenium dye N-719. A rather poor photovoltaic performance was obtained under 1 sun irradiation. (Table S1, ESI†). The bulky organic dye D35, which has shown excellent performance in combination with cobalt tris(bipyridine) redox mediators,12 was subsequently chosen as a sensitizer for the experiments in this work. The photovoltaic performance of DSCs using electrolyte ET and EI in conjunction with dye D35 under different illumination conditions are listed in Table 1. Under 1 sun irradiation, the Jsc of DSCs containing the electrolyte ET is 4.29 mA cm−2, which is only approximately 1/3 of the corresponding value of electrolyte EI. Incident photon-to-current efficiency (IPCE) spectra of the D35-sensitized solar cells with the two different electrolytes are shown in Fig. 2. Electrolyte EI gives a much higher maximum IPCE value of over 80% from 400 nm to 520 nm and a broader IPCE active range in comparison to the electrolyte ET, resulting in the higher Jsc observed for this electrolyte. UV-vis spectra of these two electrolytes are shown in Fig. S3 (ESI†). It is clear that electrolyte ET has stronger light absorption in the visible light range than electrolyte EI. Light absorption by the electrolyte diminishes the light harvesting efficiency of the dye in the DSCs. Therefore, the lower Jsc for electrolyte ET can partially be ascribed to its stronger light absorption, but this effect is mainly limited to wavelengths below 500 nm.
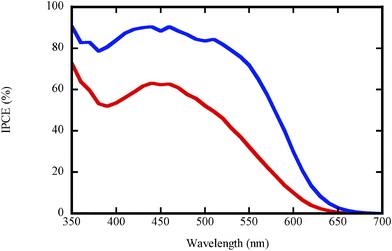 | ||
| Fig. 2 IPCE-spectra for DSCs based on electrolytes ET (red) and EI (blue). | ||
Electron injection from the excited D35 molecules into the conduction band of TiO2 is not expected to be affected by the two different electrolytes. Regeneration of the oxidized dye is, however, strongly affected by the choice of redox mediator. As is evident from the good IPCE results of D35-sensitized TiO2 solar cells, the regeneration is very efficient in case of iodide/triiodide-based electrolytes. Nanosecond laser spectroscopy was used to probe the regeneration reaction of oxidized D35 by TTF. Fig. 3 shows the transient absorption (TA) spectrum recorded in the period 0–200 ns after the laser pulse. Upon laser excitation, electrons are injected into TiO2, and oxidized D35 (D35+) is formed. In the absence of TTF, the resulting TA spectrum is due to the difference in absorption spectra of D35 and D35+, and due to the change in ground state absorption of the D35 because of a change in the electric field across the dye molecules. The latter is the Stark effect that will occur in dye-sensitized solar cells upon charge injection, as the injected electrons are charge compensated by ions in the electrolyte (or oxidized dye molecules at the surface), resulting in a change in the electric field across adsorbed dye molecules.34 At wavelengths higher than 650 nm the TA signal is free of the Stark shift, and an undisturbed absorption signal of D35+ can be observed.12 In the presence of TTF, this part of this signal disappears, demonstrating very rapid dye regeneration on the nanosecond time scale. The strong signal due to the Stark shift of the D35 is still present, which can be understood from the fact that the electric field due to electrons in TiO2 and charge-compensating ions in the electrolyte persists.
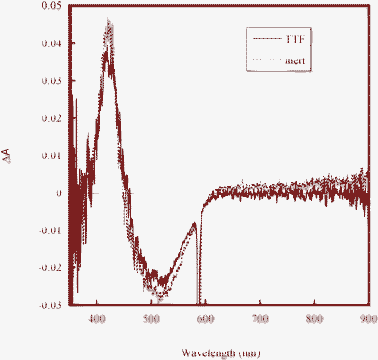 | ||
| Fig. 3 Transient absorption spectra of D35-sensitized TiO2 films in contact with an inert electrolyte and a TTF-based electrolyte. The data are collected 0–200 ns after the laser pulse. | ||
When the light intensity is gradually decreased from 1 sun to 0.1 sun, the Jsc decreases almost proportionally to the light intensities for electrolyte EI (see Table 1). In the case of electrolyte ET, however, the decrease of Jsc is not proportional to the light intensities. A higher photocurrent generation efficiency is found at lower light intensities. Such behavior could infer mass-transport limitations of the redox species in electrolyte ET. Moreover, from the EIS measurements, it is obvious that the charge-transfer resistance at the counter electrode (Rct) of the electrolyte ET is also found to be significantly higher, as shown in Fig. 4. Platinum seems not to be an optimal catalyst at the counter electrode for the charge transfer to the sulfur-containing redox electrolytes, as reported in previous publications.23,35 Therefore, the larger diffusion resistance in the electrolyte (Rdif) and Rct of electrolyte ET probably result in much lower Jsc and FF, as compared to the corresponding resistances determined for the DSCs with electrolyte EI.
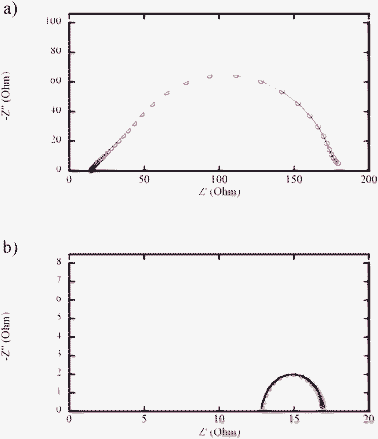 | ||
| Fig. 4 Nyquist plots of CE//electrolyte//CE systems with 0 V bias voltage: (a) electrolyte ET and (b) electrolyte EI. | ||
As described previously, DSCs containing the ruthenium dye N-719 show rather bad photovoltaic performance as compared to those containing the organic dye D35 in combination with a TTF-based electrolyte. A similar phenomenon has also been observed for the comparison between the ruthenium dye N-719 and an organic dye coded Carbz-PAHTDTT in the case of ferrocene-based electrolytes.18 The better photovoltaic performance noted for the organic dye D35 in combination with TTF-based electrolytes can most likely be attributed to the bulky alkoxy chains on the dye, which appear to suppress electron recombination loss reactions at the TiO2/dye/electrolyte interface, resulting in a better overall photovoltaic performance.12 The dye D35 was subsequently chosen as a sensitizer for the comparison between the electrolytes ET and EI. From Table 1, the Voc values for DSCs with the electrolyte ET are found to be around 200 mV lower than those of electrolyte EI at all light intensities investigated. The electron lifetime plotted as a function of the electrochemical potential of TiO2 (EF,n) is shown in Fig. S4 (ESI†). At a fixed EF,n, the electron lifetime for DSCs with electrolyte ET is considerably lower than for DSCs with electrolyte EI. At a given extracted charge (Qoc), a similar trend for the electron lifetimes was found, as shown in Fig. S5 (ESI†). This implies that the oxidized redox component in electrolyte ET recombines much faster with the photoinjected electrons at TiO2/dye/electrolyte interface, even though the alkoxy chains on the dye D35 are expected to retard the electron recombination reactions at the TiO2/dye/electrolyte interface. The rapid electron recombination results in the much lower Voc observed in spite of the fact that the redox potential of the electrolyte ET is slightly more positive than that for I−/I3− system. This is in strong accordance with results observed for other one-electron redox systems.10,12,16,18,20
Conclusions
In conclusion, the one-electron and sulfur-based TTF/TTF+ redox system has been employed and studied as an organic iodine-free redox mediator in electrolytes for DSCs. The effect of this redox mediator on the photovoltaic performance and recombination behavior was investigated and compared to the standard I−/I3− system. Cyclic voltammetry measurements show that TTF may undergo two consecutive oxidation processes, and the first potential, TTF/TTF+, was inferred to be the active process in the systems tested. A much lower open-circuit voltage was observed for electrolytes containing TTF, although the redox potential of TTF is slightly more positive than that of the I−/I3−redox couple. Electron lifetime measurements showed that the recombination between the electrons in the TiO2 film and TTF+ radical cation is much faster than that to I3−. Rather low short-circuit currents were also found for electrolytes containing TTF, which probably could be attributed to the stronger visible light absorption of TTF, combined with higher diffusion resistance in the electrolyte, as well as larger charge resistance at the counter electrode. All these properties show that the TTF system works less well than the I−/I3− system in terms of photovoltaic performance. The TTF system interestingly enough represents a sulfur-based system that is not a two-electron system, showing that recombination losses mainly can be attributed to the one-electron process than specific interactions. Also, the fact that a platinized counter electrode is a bad option for a sulfur-based redox couple is highlighted. We hope that the lessons we learned from this piece of work will shed some light on the efforts of identifying a more efficient and stable, iodine-free, alternative redox mediator in the future.Acknowledgements
We acknowledge the Swedish National Research Council, Swedish Energy Agency, the Knut and Alice Wallenberg Foundation as well as the Chinese Scholarship Council (CSC) for financial support of this work.References
- B. O'regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef CAS
.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS
- Z. Ning, Y. Fu and H. Tian, Energy Environ. Sci., 2010, 3, 1170–1181 CAS
- Z. Yu, N. Vlachopoulos, M. Gorlov and L. Kloo, Dalton Trans., 2011, 40, 10289–10303 RSC
- Y. Chiba, A. Islam, Y. Watanabe, R. Komiya, N. Koide and L. Y. Han, Jpn. J. Appl. Phys., 2006, 45, L638–L640 CrossRef CAS
- F. Gao, Y. Wang, D. Shi, J. Zhang, M. Wang, X. Jing, R. Humphry-Baker, P. Wang, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2008, 130, 10720–10728 CrossRef CAS
- H. Nusbaumer, J.-E. Moser, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. B, 2001, 105, 10461–10464 CrossRef CAS
- S. A. Sapp, C. M. Elliott, C. Contado, S. Caramori and C. A. Bignozzi, J. Am. Chem. Soc., 2002, 124, 11215–11222 CrossRef CAS
- S. Hattori, Y. Wada, S. Yanagida and S. Fukuzumi, J. Am. Chem. Soc., 2005, 127, 9648–9654 CrossRef CAS
- T. C. Li, A. M. Spokoyny, C. She, O. K. Farha, C. A. Mirkin, T. J. Marks and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 4580–4582 CrossRef CAS
- S. Yanagida, Y. Yu and K. Manseki, Acc. Chem. Res., 2009, 42, 1827–1838 CrossRef CAS
- S. M. Feldt, E. A. Gibson, E. Gabrielsson, L. Sun, G. Boschloo and A. Hagfeldt, J. Am. Chem. Soc., 2010, 132, 16714–16724 CrossRef CAS
- B. A. Gregg, F. Pichot, S. Ferrere and C. L. Fields, J. Phys. Chem. B, 2001, 105, 1422–1429 CrossRef CAS
- G. Oskam, B. V. Bergeron, G. J. Meyer and P. C. Searson, J. Phys. Chem. B, 2001, 105, 6867–6873 CrossRef CAS
- P. Wang, S. M. Zakeeruddin, J.-E. Moser, R. Humphry-Baker and M. Grätzel, J. Am. Chem. Soc., 2004, 126, 7164–7165 CrossRef CAS
- Z. Zhang, P. Chen, T. N. Murakami, S. M. Zakeeruddin and M. Grätzel, Adv. Funct. Mater., 2008, 18, 341–346 CrossRef CAS
- G. Boschloo and A. Hagfeldt, Acc. Chem. Res., 2009, 42, 1819–1826 CrossRef CAS
- T. Daeneke, T. H. Kwon, A. B. Holmes, N. W. Duffy, U. Bach and L. Spiccia, Nat. Chem., 2011, 3, 211–215 CrossRef CAS
- Y. Bai, Q. Yu, N. Cai, Y. Wang, M. Zhang and P. Wang, Chem. Commun., 2011, 47, 4376–4378 RSC
- H. N. Tsao, C. Yi, T. Moehl, J.-H. Yum, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Grätzel, ChemSusChem, 2011, 4, 591–594 CrossRef CAS
- M. K. Wang, N. Chamberland, L. Breau, J. E. Moser, R. Humphry-Baker, B. Marsan, S. M. Zakeeruddin and M. Grätzel, Nat. Chem., 2010, 2, 385–389 CrossRef CAS
- D. Li, H. Li, Y. Luo, K. Li, Q. Meng, M. Armand and L. Chen, Adv. Funct. Mater., 2010, 20, 3358–3365 CrossRef CAS
- H. Tian, X. Jiang, Z. Yu, L. Kloo, A. Hagfeldt and L. Sun, Angew. Chem., Int. Ed., 2010, 49, 7328–7331 CrossRef CAS
- J. O. Jeppesen, M. B. Nielsen and J. Becher, Chem. Rev., 2004, 104, 5115–5132 CrossRef CAS
- S. Nishida, Y. Morita, K. Fukui, K. Sato, D. Shiomi, T. Takui and K. Nakasuji, Angew. Chem., Int. Ed., 2005, 44, 7277–7280 CrossRef CAS
- S. Wenger, P.-A. Bouit, Q. Chen, J. Teuscher, D. D. Censo, R. Humphry-Baker, J.-E. Moser, J. L. Delgado, N. Martín, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2010, 132, 5164–5169 CrossRef CAS
- A. J. Olaya, P. Ge, J. F. Gonthier, P. Pechy, C. Corminboeuf and H. H. Girault, J. Am. Chem. Soc., 2011, 133, 12115–12123 CrossRef CAS
- D. Canevet, M. Salle, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245–2269 RSC
- Z. Zhang, Enhancing the open-circuit voltage of Dye-sensitized solar cells: coadsorbents and alternative redox couples, PhD thesis, EPFL, 2008 Search PubMed
- D. P. Hagberg, X. Jiang, E. Gabrielsson, M. Linder, T. Marinado, T. Brinck, A. Hagfeldt and L. C. Sun, J. Mater. Chem., 2009, 19, 7232–7238 RSC
- Z. Yu, M. Gorlov, J. Nissfolk, G. Boschloo and L. Kloo, J. Phys. Chem. C, 2010, 114, 10612–10620 CAS
- G. Boschloo, L. Haggman and A. Hagfeldt, J. Phys. Chem. B, 2006, 110, 13144–13150 CrossRef CAS
- H. Tian, X. Yang, R. Chen, Y. Pan, L. Li, A. Hagfeldt and L. Sun, Chem. Commun., 2007, 3741–3743 RSC
- U. B. Cappel, S. M. Feldt, J. Schöneboom, A. Hagfeldt and G. Boschloo, J. Am. Chem. Soc., 2010, 132, 9096–9101 CrossRef CAS
- H. Tian, Z. Yu, A. Hagfeldt, L. Kloo and L. Sun, J. Am. Chem. Soc., 2011, 133, 9413–9422 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ra00877c |
| This journal is © The Royal Society of Chemistry 2012 |