Naphthalene-dioxygenase catalysed cis-dihydroxylation of bicyclic azaarenes†
Claude
Chopard
*a,
Gildas
Bertho
a and
Thierry
Prangé
b
aLaboratoire de Chimie et Biochimie Pharmacologiques et Toxicologiques, UMR 8601 CNRS, Université Paris Descartes, 45 rue des Sts Pères, 75270, Paris, cedex 06, France. E-mail: mailto:claude.chopard@parisdescartes.fr; Tel: +33 1 42 86 21 75
bLaboratoire de Cristallographie et RMN Biologiques, UMR 8015 CNRS, Université Paris Descartes, 4 avenue de l'Observatoire, 75270, Paris, cedex 06, France
First published on 15th November 2011
Abstract
The scope of the biotransformation of a series of azaarene compounds by recombinant whole-cells of Escherichia coli JM109(DE3)(pDTG141) expressing the naphthalene dioxygenase system (NDOS) from Pseudomonas sp. NCIB 9616-4 was explored. The present study establishes that several bicyclic azaarenes are good substrates in the NDO-catalysed reaction, giving cis-dihydrodiol derivatives; monohydroxylated products were also observed.
1. Introduction
The bacterial metabolism of aromatic compounds often proceed via an initial dihydroxylation catalyzed by Rieske non-heme iron dioxygenases to yield cis-dihydrodiol derivatives. These multicomponent bacterial systems, mainly toluene dioxygenase (TDOS), naphthalene dioxygenase (NDOS) and biphenyl dioxygenase (BPDOS), have been frequently utilized to produce, by environmentally friendly routes, hundreds of such cis-1,2-dihydrodiols, several of which have been used as chiral synthons for the synthesis of biologically active products and value-added chemicals.1–3In recent years, direct evidence for the cis-dihydroxylation of a 2-pyridone ring by NDO has been reported.4–6 A major dihydroxylated metabolite, (5S,6S)-5,6-dihydroxy-1-methyl-5,6-dihydropyridin-2(1H)-one was obtained from the corresponding N-methyl-2-pyridone substrate, together with small amounts of its 3,4-dihydroxy isomer when E.coli JM109(DE3)(pDTG141) expressing the NDO system from Pseudomonas sp. NCIB 9616-4 (a strain developed by Gibson7) was used as a whole-cell biocatalyst. We have also shown that 1-methyl quinolone was transformed by this strain more efficiently than 1-methyl pyridone (62% transformation vs. 35–40%); probably due to a better accommodation of 1-methyl quinolone by the active site of NDO. Azaheterocyclic cis-dihydrodiols with a high enantiomeric purity are formed by this bacterial dioxygenase and their absolute configuration was directly and definitively determined.6 The effect of substituents on 2-pyridone and 2-quinolone rings in the NDO-catalysed reaction was also investigated.
The cis-dihydroxylation of bicyclic fused azaarenes (quinoline, isoquinoline, quinoxaline, quinazoline, substituted quinolines) has been reported, using the multicomponent bacterial toluene dioxygenase (TDOS).8–10 More recently Boyd et al. have compared different types of dioxygenase enzymes (TDO, NDO and BPDO) for the regio- and stereo-selectivity of the cis-dihydroxylation of several monocyclic azaarene analogues to biphenyl and tricyclic azaarenes, showing that the larger active site of BPDO was more efficient to accommodate sterically demanding tricyclic azaphenantrenes.11
Azaarenes (often quoted as NPAHs) are widely distributed throughout the global environment where they are persistent. Some compounds of this group exert toxic effects.12 Quinoline is one of the most abundant and has been shown to be both carcinogenic and mutagenic.13,14 Bioremediation is one of the strategies developed to rehabilitate polluted environments.15,16 Several bacterial strains are able to metabolise some polycyclic azaarenes.17 Among them, Pseudomonas putida UV4, which expressed toluene dioxygenase (TDO),9,10 has been shown to transform quinoline and other bicyclic azaarenes, but in a very low extent perhaps because of its better specificity toward monocyclic arenes. Nevertheless, higher yields were reported when using another (unspecified) strain of P. putida.8 As quinoline is almost isosteric to naphthalene, a bacterial strain expressing naphthalene dioxygenase should be a better choice. NDO-catalysed biotransformation of fused bicyclic azaarenes should be expected to occur in good yields, providing less toxic compounds as metabolites. From another point of view, quinoline or isoquinoline moieties are present in many classes of biologically-active compounds. New metabolites could give access to modifications of the quinoline or isoquinoline moieties which can trigger interesting biological activities.
In this study we have used E.coli JM109(DE3)(pDTG141) overexpressing the NDO system from Pseudomonas sp. NCIB 9616-4 for the oxidative biotransformation of fused bicyclic azaarenes reported in Scheme 1.
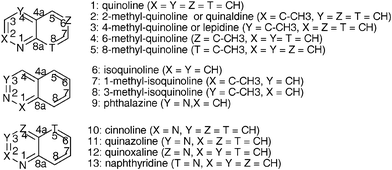 | ||
| Scheme 1 Azaarenes used as substrates for the biotransformations and numbering. | ||
2. Results
The previously used E. coli JM109(DE3)(pDTG141), a strain that overexpresses the NDO system from Pseudomonas sp. NCIB 9816-4, was used as a whole-cell biocatalyst. Biotransformation of each of the compounds (1–13) was conducted in the experimental conditions precedently optimised for 1-methyl pyridone.6 HPLC-ESIMS analysis (positive mode) of aliquots of the incubation mixtures withdrawn at intervals shows the initial substrate (M + H+), dihydrodiol(s) (M + 34 + H+) and/or monohydroxylated derivative(s) (M + 16 + H+) (see Table 1 and 2). Conversion yields were evaluated by comparing the integrated mass peak areas of the products to that of remaining substrate. The initial conversion rate remained in a lower range (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2–1:60) compared to naphthalene dihydroxylation in the same conditions. The conversion yield (after 30 h) was in the 20–100% range (see Tables 1 and 2). Preparative experiments were then conducted with each substrate. The supernatants of incubation media were lyophilized and extraction with aceton of the dry powder gave the crude organic extracts. The oxidation products were isolated from the crude organic extract by flash chromatography on silicagel or a prepacked C18 column, and characterised by NMR analysis. The relative yield of isolated products was estimated from the 1H NMR spectra of the crude organic extracts using integration of representative signals.
:2–1:60) compared to naphthalene dihydroxylation in the same conditions. The conversion yield (after 30 h) was in the 20–100% range (see Tables 1 and 2). Preparative experiments were then conducted with each substrate. The supernatants of incubation media were lyophilized and extraction with aceton of the dry powder gave the crude organic extracts. The oxidation products were isolated from the crude organic extract by flash chromatography on silicagel or a prepacked C18 column, and characterised by NMR analysis. The relative yield of isolated products was estimated from the 1H NMR spectra of the crude organic extracts using integration of representative signals.
| Substrate | Total conversion (%) and relative yield of isolated products (%) | m/z expected | Identified major products | ||
|---|---|---|---|---|---|
| NDOa | TDOb | P. putida (mutant)c | |||
| a This work, E. Coli JM109(DE3)(pDTG141). b From ref. 9, P. putida UV4. c From ref. 8, mutant of P. putida. | |||||
| 66 | <10 | 100 | |||
 1 (Quinoline)
1 (Quinoline) |
130 | Quinoline | |||
| 55 | 33 | 33 | 164 | 5R,6S-Dihydrodiol (1d1) | |
| 35 | 13 | 13 | 146 | Quinolin-3-ol (1h3) | |
| 6 | n.d. | n.d. | 164 | cis-7,8-Dihydrodiol (1d2) | |
| 4 | 27 | 27 | 146 | Quinolin-8-ol (1h8) | |
| 0 | 27 | 27 | 136 (ESI-) | Anthranilic acid | |
 2 (2-Methylquinoline)
2 (2-Methylquinoline) |
>99 | ||||
| 144 | 2-Methylquinoline | ||||
| 92 | 178 | 5R,6S-Dihydrodiol (2d1) | |||
| 8 | 160 | 2-Methylquinolin-8-ol (2h8) | |||
 3 (4-Methylquinoline)
3 (4-Methylquinoline) |
20 | ||||
| 144 | 4-Methylquinoline | ||||
| 80 | 160 | Quinolin-4-ylmethanol (3e) | |||
| 20 | 178 | cis-5,6-Dihydrodiol (3d1) | |||
| Quinolin-4-ylmethanol | |||||
 4 (6-Methylquinoline)
4 (6-Methylquinoline) |
45 | ||||
| 144 | 6-Methylquinoline | ||||
| 100 | 160 | 6-Methylquinolin-3-ol (4h3) | |||
 5 (8-Methylquinoline)
5 (8-Methylquinoline) |
78 | ||||
| 144 | 8-Methylquinoline | ||||
| 72 | 160 | Quinolin-8-ylmethanol (5e) | |||
| 28 | 178 | cis-5,6-Dihydrodiol (5d1) | |||
| 74 | 5 | 100 | |||
 6 (Isoquinoline)
6 (Isoquinoline) |
130 | Isoquinoline | |||
| 60–65 | 42 | 45 | 146 | Isoquinolin-5-ol (6h5) | |
| 16 | 58 | 47 | 164 | cis-7,8-Dihydrodiol (6d2) | |
| 6–8 | n.d | 164 | cis-5,6-Dihydrodiol (6d1) | ||
| 16 | n.d. | n.d. | 146 | Isoquinolin-4-ol (6h4) | |
| 0 | n.d. | n.d. | 146 | Isoquinolin-8-ol and -1-ol | |
 7 (1-Methylisoquinoline)
7 (1-Methylisoquinoline) |
52 | ||||
| 144 | 1-Methylisoquinoline | ||||
| 71 | 160 | Isoquinolin-1-ylmethanol (7e) | |||
| 29 | 178 | 7S,8R-Dihydrodiol (7d2) | |||
 8 (3-Methylisoquinoline)
8 (3-Methylisoquinoline) |
>99 | ||||
| 144 | 3-Methylisoquinoline | ||||
| 73 | 178 | cis-7,8-Dihydrodiol (8d2) | |||
| 27 | 160 | 3-Methylisoquinolin-5-ol (8h3) | |||
| Substrate | Total conversion (%) and relative yield of products (%) | m/z expected | Identified major products | ||
|---|---|---|---|---|---|
| NDOa | TDOb | P. putida (mutant)c | |||
| a This work, E. Coli JM109(DE3)(pDTG141). b From ref. 9, P. putida UV4. c From ref. 8, mutant of P. putida. | |||||
 9 (Phthalazine)
9 (Phthalazine) |
20 | ||||
| 131 | Phthalazine | ||||
| 10 | 165 | Dihydrodiol | |||
| 90 | 147 | Phthalazin-5-ol (9h5) | |||
 10 (Cinnoline)
10 (Cinnoline) |
25 | ||||
| 131 | Cinnoline | ||||
| n.d | 165 | Dihydrodiol | |||
| n.d. | 147 | Cinnolinols | |||
| >99 | <10 | 100 | |||
| 131 | Quinazoline | ||||
 11 (Quinazoline)
11 (Quinazoline) |
56 | 55 | 55 | 165 | 5R,6S-cis-Dihydrodiol (11d1) |
| 0 | 167 | 5,6-cis-Tetrahydrodiol | |||
| 0 | 45 | 10 | 147 | Quinazolin-4-ol | |
| 43 | — | — | 147 | Quinazolin-8-ol (11h8) | |
 12 (Quinoxaline)
12 (Quinoxaline) |
>99 | <10 | 100 | ||
| 131 | Quinoxaline | ||||
| 100 | 47 | 45 | 165 | 5R,6S-cis-Dihydrodiol (12d1) | |
| 0 | 44 | 40 | 147 | Quinoxalin-5ol | |
| 0 | 9 | 20 | 147 | Quinoxalin-2ol | |
 13 (1,5-Naphthyridine)
13 (1,5-Naphthyridine) |
20 | ||||
| 131 | 1,5-Naphthyridine | ||||
| n.d. | 165 | Dihydrodiol | |||
| n.d. | 147 | Naphthyridinols | |||
2.1 Quinoline, isoquinoline and C-methyl derivatives
Concerning the regioselectivity of the reaction, assuming a molecular similarity with naphthalene, two different cis-dihydrodiols were expected on the carbo ring; the 5,6-position is favoured (55% yield) compared to the 7,8-position, even if the small amount of quinolin-8-ol (1h8) formed may come from a spontaneous dehydration of the 7,8-dihydrodiol (1d2) (10% yield). For the aza ring two positions for dioxygenation should be considered. As expected, due to nitrogen in position 1, no transformation on the 1 and/or 2 position was observed. The cis-3,4-dihydrodiol was not isolated, but its spontaneous dehydration could produce the quinolin-3-ol (1h3) formed in 35% yield. The optical rotation of the purified 5,6-dihydrodiol 1d1 ([α]D +204; c 0.032, MeOH) shows that it is a single enantiomer, identical to that previously isolated,9 (5R,6S)-5,6-dihydroquinoline-5,6-diol. The oxidation is not specific of the carbo ring of quinoline.
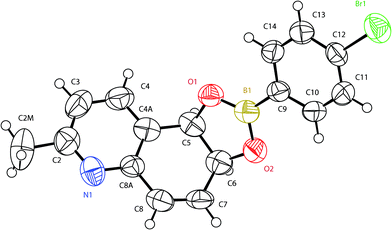 | ||
| Fig. 1 X-Ray structure of the 4-bromophenylboronate derivative of (5R,6S)-2-methyl-5,6-dihydroquinoline-5,6-diol (2d1B). | ||
Analysis of the structure (Fig. 1) unequivocally revealed a cis-diaxial substitution with an absolute (5R,6S) configuration based on the clear indication of the Flack index at the end refinements.18,19 Put together with the high positive value of the optical activity, this also suggested that the dihydrodiol 2d1 was the enantiopure (5R,6S)-2-methyl-5,6-dihydroquinoline-5,6-diol.
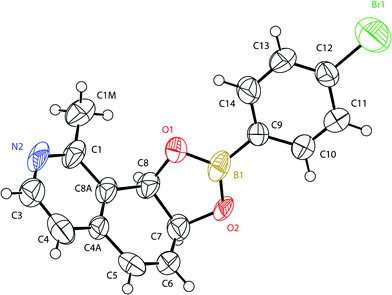 | ||
| Fig. 2 X-Ray structure of the 4-bromophenylboronate derivative of (7S,8R)-1-methyl-7,8-dihydroisoquinoline-7,8-diol (7d2B). | ||
As for the derivative of 2d1B, the crystal structure analysis (Fig. 2) revealed a cis-diaxial derivative, with the (7S, 8R)-absolute configuration, based on the anomalous scattering of the bromine atom. This suggested that dihydrodiol 7d2 was the enantiopure (7S,8R)-1-methyl-7,8-dihydroisoquinoline-7,8-diol.
The results obtained for the biotransformations of compounds 1–8 are summarized in Table 1.
2.2 Bis-azaarenes
The results obtained for the biotransformation of compounds 9–13 are summarized in Table 2.
3. Discussion
The substrates tested (1–13) are transformed by E. coli JM109(DE3)(pDTG141) into one or several oxidized products with the following efficiencies: quinazoline (11) > 2-methylquinoline (quinaldine) (2), 3-methylisoquinoline (8), 8-methylquinoline (5), quinoxaline (12) > isoquinoline (6), quinoline (1), 1-methylisoquinoline (7) > 6-methylquinoline (4) > 4-methylquinoline (lepidine) (3), phthalazine (9), naphthyridine (13), cinnoline (10). cis-Dihydrodiols have been detected from all of them except with 4 were only 6-methylquinolin-3-ol was obtained, and 13. The cis configuration of most of the dihydrodiols was chemically confirmed by the formation of their bromophenyl boronate derivatives which were also used for the determination of their absolute configurations by X-ray crystallography. Other monohydroxylated phenol type compounds have also been recovered in the biotransformations of 1, 2, 6, 8, 10, and 11 either resulting from the spontaneous dehydration of a primarily formed cis-dihydrodiol or directly formed via a monohydroxylation reaction.21 A third reaction type mediated in the active site results in the hydroxylation of methyl substituents, exclusively when the methyl substitution of the substrate is adjacent to the ring junction, as precedently reported for 1-methylnaphthalene.22 Referring to quinoline or isoquinoline, and depending on its position on the carbo- or aza-ring, a methyl substituent is able to affect in a large extent the biotransformation yield and the regioselectivity of the reactions as previously described for benzofurane or benzothiophene.23Regarding the dihydroxylation reaction, the published crystallographic data from Karlsson et al. of the enzyme (PDB ID: 1O7G) suggests that the positioning of a substrate toward iron in the active site can be deduced from the structure and the stereochemistry of the produced dihydrodiol(s).24 We propose a description of our and other results where we consider that a bicyclic substrate can occupy in the active site the same place as naphthalene. Due to the asymmetry of the structure of the active site including the catalytic iron, four dihydroxylated products could be obtained, each of them corresponding to a distinct orientation (or “pose”) of the substrate in the active site (referred to as A, B, C and D in Scheme 2). We have to note that the stereochemistry of the dihydrodiol is undissociable from its location on the structure. The number of products formed can be lowered due to the geometry of the substrate. With naphthalene where the four poses are identical, only one product, (1R,2S)-cis1,2-dihydro-1,2-dihydroxy-naphthalene can be recovered. Anthracene, where also the four poses are identical due to the symmetry of the molecule, similarly gives a single dihydrodiol (1R,2S)-1,2-dihydroanthracene-1,2-diol.25
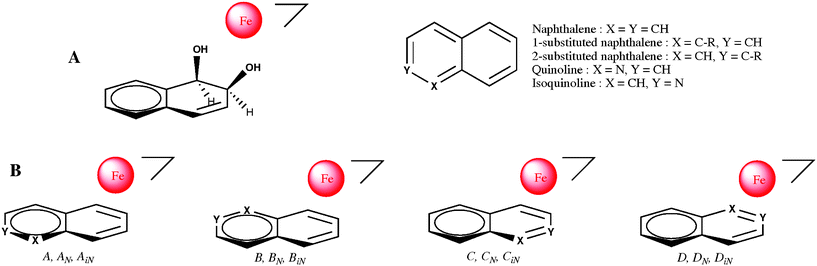 | ||
| Scheme 2 A: Relative positioning of 1,2-dihydrodihydroxy-naphthalene with respect to mononuclear iron in the active site of NDO (from crystallographic data26). B: The four possible poses for a N-monosubstituted bicyclic substrate in the active site: no subscript refers to naphthalene, N subscripts refer to quinoline and iN subscripts refer to isoquinoline. | ||
The regio- and stereochemical analysis of the products formed from a series of monosubstituted naphthalenes using P. fluorescens TTC1 (expressing NDO) have been previously performed by Bestetti et al.22 If we refer to our model of the active site (Scheme 2), the regio- and stereochemistry of the dihydrodiols they obtained for the 2-substituted naphthalenes correspond to pose B, but trace amounts of other products suggest that pose A is also adopted for 2–Cl and 2–NO2 naphthalenes. Thus for 2-substituted naphthalenes, pose B ≫ pose A > pose C, and pose D). With 1-substituted naphthalenes two dihydrodiols are usually obtained for each substituted compound corresponding to pose B and pose A (pose B > pose A). Nevertheless, with a methyl or ethyl substituent, only one dihydrodiol is formed, namely (1R,2S)-8-methyl or (1R,2S)-8-ethyl-1,2-dihydronaphthalene-1,2-diol corresponding to pose B. We have to note that no dihydrodiol is formed corresponding to the substrate in pose C or in pose D. Phenanthrene,27 and benzo(h)quinoline and benzo(f)quinoline recently studied by Boyd et al.,28 can be considered as 1,2-disubstituted naphthalenes, and should adopt pose B in the active site; accordingly, the major bioproducts obtained were respectively (3S,4R)-3,4-dihydrophenanthrene-3,4-diol, (9S,10R)-9,10-dihydrobenzo[h] quinoline-9,10-diol, and (9S,10R)-9,10-dihydrobenzo[f] quinoline-9,10-diol, corresponding to the usual regioselectivity observed for the bay region and with the expected stereospecificity. Minor cis-dihydroxylated products, quoted “abnormal” by Boyd et al. because they are formed at a non-bay region of the substrate, are also isolated in these biotransformations with NDO,28 respectively (1R,2S)-1,2-dihydrophenanthrene-1,2-diol, and (7R,8S)-7,8-dihydro-benzo[h]quinoline-7,8-diol and agree with a pose A of these substrates in the active site (pose B > pose A). The proposed scheme is validated by the structure and the stereochemistry of the dihydrodiols isolated by the different authors and the high enantiomeric excess found for these products.
In the particular case of 1-methyl naphthalene, a minor different metabolite is isolated, naphthalen-1-yl methanol formed enzymatically by benzylic oxidation, revealing for this type of reactivity a pose for the substrate close to pose D (perhaps shifted to allow the methyl group to be close enough to iron in the catalytic pocket).
From a geometrical point of view, it should be expected that quinoline (1) and isoquinoline (6) occupy the same four poses as naphthalene in the active site, but due to the presence of a nitrogen atom, they are not equivalent. The dihydrodiols obtained by biotransformation of quinoline (1) have been identified to (5R,6S)-5,6-dihydroquinoline-5,6-diol (1d1) and 7,8-dihydroquinoline-7,8-diol (1d2), which implies the two poses A and B for the substrate (further referred to as AN and BN in Scheme 2). Accordingly, the absolute configuration of (1d1) is 5R,6S, as observed by Boyd et al. in the biotransformation with TDO.9 If we consider that the absolute configuration of the 7,8-dihydrodiol (1d2) obtained with NDO is 7S,8R as previously established with TDO, then dihydroxylation occurs according with the substrate in pose BN. Assuming that quinolin-3-ol (1h3) is produced from the cis-3,4-diol by spontaneous dehydration, that means that pose C would be also allowed for the substrate (referred to as CN in Scheme 2). No hydroxylated product has been detected which could have been formed with the substrate at pose D (referred to as DN in Scheme 2). For the efficiency of the dihydroxylation reaction, we have observed that pose AN ≈ pose CN ≫ pose BN ≫ pose DN. The regio- and stereo-specificity for the 5R,6S-dihydrodiol 12d1 we observe for the dihydroxylation of quinoxaline (12), shows as expected, that the substrate occupies only pose AN, and that a pose similar to DN is not accepted.
The products identified after biotransformation of isoquinoline (6), (5R,6S)-5,6-dihydroisoquinoline-5,6-diol (6d1) (and isoquinolin-5-ol (6h5)), cis-7,8-dihydroisoquinoline-7,8-diol and isoquinolin-4-ol suggest that the allowed poses for the substrate are also respectively AN, BN and CN (referred to as AiN , BiN and CiN in Scheme 2), without any product corresponding to dihydroxylation in pose DN (referred to as DiN in Scheme 2). For the efficiency of the dihydroxylation reaction, we have obtained pose AiN ≫ pose BiN > pose CiN ≫ DiN.
For quinoline and isoquinoline, specific interactions of nitrogen with the active site could be involved for the positioning of the substrate favouring poses AN and AiN , and excluding poses DN and DiN for cis-dihydroxylation reaction. Quinazoline (11) can be considered either as a quinoline or as an isoquinoline. The products obtained, the 5,6-dihydrodiol 11d1 and quinazolin-8-ol (11h8) are those expected from each of the preferred poses (pose AN and pose AiN) of quinoline and isoquinoline respectively. Therefore the stereochemistry of 11d1 should be 5R,6S as also suggested by the high positive value of the optical activity.
For methyl substituted quinolines and isoquinolines the structure of the dihydrodiol formed is due to the position of the methyl substituent (as with substituted naphthalene) and to the position of nitrogen. In the case of quinaldine (2), the dihydrodiol expected from a major role of nitrogen (pose AN) would be the 5R,6S-dihydrodiol while the effect of the methyl substituent would favour the 7S,8R-dihydrodiol (pose B). As the major product obtained is the 5R,6S-dihydrodiol (2d1), as expected for a director role of nitrogen in the positioning of this substrate at the active site (pose AN). With lepidine (3) and 8-methylquinoline (5), the regioselective formation of the 5,6-dihydrodiols 3d1 and 5d1 is observed (pose AN) but the major way of transformation is the benzylic oxidation of the methyl substituent (in pose D), which occurs as a minor way with 1-methyl-naphthalene. For 6-methylquinoline (4), the 3-hydroxy derivative 4h3 may be produced by direct monohydroxylation of the substrate or by the spontaneous dehydration of the 3,4-dihydrodiol. Therefore the substrate should be in pose B (as 2-methylnaphthalene) corresponding to pose CN of quinoline and excluding pose AN. For 1-methylisoquinoline (7) the 7S,8R cis-dihydrodiol obtained (7d2) is not that expected from an isoquinoline (pose AiN) but from 1-methylnaphthalene (pose B). The low value of its αD prompted us to investigate its chirality by CD spectrum, versus the cis-dihydrodiol from naphtalene used as a reference.29 The same Cotton effect was observed suggesting equivalent absolute configurations and hence favoring pose B in the active site. The absolute configuration was definitely proven by X-ray diffraction (Fig. 2). In addition, the benzylic oxidation of the methyl substituent also occurs (pose D). Biotransformation of 3-methylisoquinoline (8) is one of the most efficient in this series of compounds. No 5,6-dihydrodiol is observed and 5-hydroxy derivative is only a minor product whereas, in the pose AiN for isoquinoline or pose B for 2-methylnaphthalene, such hydroxylation should be favoured. The formation of cis-7,8-dihydrodiol (8d2) as the major product is not expected and suggests a specific interaction within the active site for the methyl substituent of 8 in pose A. Thus the stereochemistry of the cis-7,8-dihydrodiol of 3-methylisoquinoline (8d2) should be 7S,8R, as suggested also by the large positive value of the optical activity of this compound.
From this description of our results we can note for the regiospecific dihydroxylation of isoquinoline and C-methyl derivatives of quinolines (excepted for 6-methylquinoline), better efficiency is observed with substrate in poses AN and AiN, pose AN ≈ pose CN for quinoline, pose CN for 6-methyl quinoline, and pose BiN for C-methyl derivatives of isoquinolines.
We have also observed an enhanced efficiency of dihydroxylation of quinaldine (2) compared to that of quinoline (1) put together with the unexpected formation of the cis-7,8-dihydrodiol (8d2) as the major product of oxidation of 3-methylisoquinoline (8) this suggests a specific interaction within the active site for the methyl substituent of these substrates in pose A. It would be interesting to identify the amino acid(s) of the active site involved in this interaction and substrate docking simulations would be helpful.
Benzylic hydroxylation is the major way of biotransformation for 3, 5, and 7. This reaction implies pose D for the substrate and is not dependent of the location of nitrogen on the bicyclic structure. As for 1-methylnaphthalene, a regiospecific cis-dihydroxylation is in competition with this hydroxylation.
4. Conclusion
The results of the present investigation clearly show that quinoline, isoquinoline and their frequently associated methyl derivatives as environmental pollutants (often quoted as NPAHs) are transformed by the whole cells of E. coli JM109(DE3)(pDTG141) expressing the naphthalene dioxygenase system from Pseudomonas sp. NCIB 9616-4. Optically active cis-dihydrodiols and/or the corresponding phenol derivatives of the carbocycle are generally produced. Nevertheless, it appears also that when the methyl substituent is at the vicinity of the junction of the bicyclic structure its monohydroxylation occurs preferentially (3, 5 and 7), independent of the nitrogen position. An important set of new products with valuable stereochemical and/or functional properties was generated by these biotransformations.5. Experimental
5.1 Substrates and chemicals
Substrates were from commercial origin : quinoline, quinaldine, 1-methylisoquinoline, 3-methylisoquinoline, cinnoline hydrochloride, and naphthyridine hydrochloride from Aldrich, lepidine, isoquinoline, 6-methylquinoline, phthalazine, quinoxaline, and quinazoline, from Acros Organics and, 8-methylquinoline from Lancaster, and used without further purification except quinoline which was distilled before use.5.2 Analytical procedures
1H NMR and 13C NMR spectra were recorded in (CD3)2CO (unless stated otherwise) on Bruker ARX250 (250 MHz for 1H) or Bruker Avance500 (500 MHz for 1H and 125 MHz for 13C) spectrometers. Chemical shifts (δ) are reported in ppm, and coupling constants are given in Hz. 1H signals were assigned using 2D homonuclear 1H-1H COSY and TOCSY experiments. The TOCSY and the COSY spectra were used to distinguish the spin systems and to confirm assignments of the hydrogen resonances. The 3J1H,1H coupling constants were determined by homodecoupling experiments of 1D 1H spectra. 13C signals were assigned using 2D heteronuclear 1H-13C HSQC and HMBC experiments. Additionally, 2D-NOESY experiments were used to assign the spin systems closely related. Optical rotation measurements ([α]D) were given in 10−1 deg cm2 g−1, and were carried out with a Perkin Elmer 241 polarimeter at ambient temperature (ca. 20 °C). HPLC-ESIMS analyses were performed with a Thermo Finnigan LCQ Advantage Instrument (electrospray positive mode) equipped for HPLC with a Polarity dC18 column (waters, 4.6 × 250 mm, 5 μm) equilibrated with the appropriate water–CH3CN mobile phase. Products were eluted with a CH3CN gradient containing formic acid (999:1) at 40 °C and 0.4 cm3 min−1 flow rate. Purification of the organic extract of biotransformation was achieved by SP-flash chromatography on Kieselgel Si60 (Merck, 40–63 μm) columns equilibrated with CH2Cl2 and eluted with CH2Cl2/(CH3)2CO/CH3OH in the appropriate ratios and reversed-phase (RP)-flash chromatography over a prepacked C18 column (7.5 cm × 1.5 cm; AIT, Houilles, France), using a gradient from 0/100 to 60/40 of CH3CN/H2O mixture. The CD spectra were recorded on a JASCO J-815 spectrometer in methanol solutions. The two X-ray structure coordinates of 2d1B and 7d2B were deposited with the Cambridge Crystallographic Data Centre (CCDC) 12 Union road, Cambridge CB2 1EZ, UK under reference codes 815425 and 815426, respectively, they are available upon request to http://www.ccdc.cam.ac.uk/.
5.3 Micro organism and growth conditions
Escherichia coli JM109(DE3)(pDTG141) was kindly provided by D. T. Gibson (Iowa University). Cells were grown as previously described,6 and their dihydroxylating activity toward naphthalene routinely examined in initial rate conditions as described for analytical biotransformations (see below). Activity was found to be about 1–2 mM 1,2-dihydroxydihydronaphthalene formed/hour/OD600 unit.5.4 General procedure for analytical biotransformations
In optimised conditions, frozen cells were suspended in 20 cm3 of 0.1 M phosphate buffer pH 7.2 (0.5 g of cells, OD600 about 7), containing sodium ampicillin (0.27 mM, 0.1 g L−1) and glucose (0.2%) in 125 cm3 conical flasks. After addition of 5 mg of substrate, the biotransformation was conducted at 27 °C with orbital shaking at 250 rpm for 30 h. Aliquots (0.7 cm3) were withdrawn at intervals, centrifuged and micro filtered for direct HPLC-MS analysis.5.5 General procedure for preparative biotransformations
Frozen cells (about 12 g) were resuspended in 500 cm3 of 0.1 M phosphate buffer (pH 7.2) containing sodium ampicillin (0.27 mM) and glucose (0.2%) in 2 L conical flasks. After addition of 150 mg of substrate, biotransformations were conducted at 27 °C with orbital shaking at 250 rpm for 30 h. Aliquots (0.7 cm3) were withdrawn at intervals, centrifuged and micro filtered for direct HPLC-MS analysis.5.6 Characterization and identification of the bioproducts
Two products were isolated by flash chromatography of preparative experiments and identified as cis-2-methyl-5,6-dihydroquinoline-5,6-diol (2d1) and 2-methylquinolin-8-ol (2h8) by 1H and 13C NMR analysis.
cis-2-Methyl-5,6-dihydroquinoline-5,6-diol (2d1). δ H (500 MHz, (CD3)2CO): 7.05 (1H, d, J3–4 = 7.6, 3-H), 7.68 (1H, d, J4–3 = 7.6, 4-H), 4.64 (1H, d, J5–6 = 4.9, 5-H), 4.30 (1H, t, J = 4.9, 6-H), 6.27 (1H, dd, J7–8 = 9.9, J7–6 = 4.3 7-H), 6.52 (1H, d, J8–7 = 9.9, 8-H), 2.42 (3H, s, CH3); δC (125 MHz, (CD3)2CO): 157.3 (C2), 122.1(C3), 135.7 (C4), 130.5 (C4a), 70.5 (C5), 67.2 (C6), 134.7 (C7), 131.0 (C8), 151.7 (C8a), 23.8 (CH3). Optical rotation measurement of 2d1 ([αD]) was + 140 (c 0.028, MeOH).
(4-Bromo)phenylboronate derivative (2d1B). 2d1 (85 mg, 0.48 mmol) and 4-bromophenyl boronic acid (105 mg, 1.1 equiv.) were reacted in CH2Cl2/(CH3)2CO (5 cm3) 1/1 at room temperature during 1 h. The product formed was purified by flash chromatography on silica with CH2Cl2 as eluent. δH (250 MHz, CDCl3): 7.82 (1H, d, J4–3 = 7.8, 3-H), 7.21 (1H, d, J4–3 = 7.8, 4-H), 6.63 (1H, d, J6–5 = 10.0, 5-H), 6.30 (1H, dd, J6–5 = 10.0, J6–7 = 3.3, 6-H), 5.56 (1H, ddd, J8–7 = 7.8, J7–6 = 3.3, 4J7–5 = 1.2, 7-H), 5.81 (1H, d, J8–7 = 8.3, 8-H), 2.47 (3H, s, CH3), 7.62–7.75 (4H, m, Ar–H). Crystals suitable for X-ray analysis were grown by slow evaporation from a CH2Cl2 solution as thin prisms, they were recorded on a Bruker AXS CCD diffractometer (Mo-Kα wavelength). Crystal data for 2d1B: monoclinic, P21, with parameters: a = 4.320(5); b = 10.637(4); c = 16.090(4) Å; β = 94.11(5)° and Z = 2. Structure was refined to R = 3.7% (1495 observed Fo) and R = 5.3% (all 1799 Fo data).
2-Methylquinolin-8-ol (2h8). δ H (250 MHz, (CD3)2CO): 8.21 (1H, d, J4–3 = 8.5, 4-H), 7.46 (1H, d, J3–4 = 8.5, 3-H), 7.42 (1H, t, J5–6 = 7.8, 6-H), 7.38 (1H, d, J5–6 = 7.8, 5-H), 7.11 (1H, d, J7–6 = 7.8, 7-H), 2.72 (3H, s, CH3); δC (125 MHz, (CD3)2CO): 159.1 (C2), 124.8 (C3), 138.3 (C4), 128.9 (C4a), 119.6 (C5), 128.7 (C6), 111.9 (C7), 154.3 (C8), 139.8 (C8a), 26.1 (CH3).
Two products were isolated by flash chromatography of preparative biotransformations and identified as quinolin-4-ylmethanol (3e) and cis-4-methyl-5,6-dihydroquinoline-5,6-diol (3d1) by 1H and 13C NMR spectroscopy.
Quinolin-4-ylmethanol (3e). δ H (500 MHz, (CD3)2CO): 8.92 (1H, d, J2–3 = 3.8, 2-H), 7.68 (1H, d, J3–2 = 4.4, 3-H), 7.63 (1H, t, J6–5 = 7.6, 6-H), 7.78 (1H, t, J7–8 = 7.6, 7-H), 8.11 (2H, t, J = 7.1, 5-H and 8-H), 5.24 (2H, s, CH2); δC (125 MHz, (CD3)2CO): 152.0 (C2), 119.7 (C3), 149.6 (C4), 127.6 (C4a), 131.4 (C5), 128.0 (C6), 130.6 (C7), 131.6 (C8), 149.4 (C8a), 62.0 (CH2).
cis-4-Methyl-5,6-dihydroquinoline-5,6-diol (3d1). δ H (500 MHz, (CD3)2CO): 8.25 (1H, d, J2–3 = 5.2, 2-H), 7.02 (1H, d, J3–2 = 5.2, 3-H), 4.84 (1H, dd, J5–6 = 5.2, 4J5–7 = 1.5, 5-H), 4.52 (1H, m, 6-H), 6.09 (1H, dt, J7–8 = 9.8, 4J7–5 = 1.8, 7-H), 6.43 (1H, dd, J8–7 = 10.0, 4J8–6 = 2.7, 8-H), 2.41 (3H, s, CH3); δC (125 MHz, (CD3)2CO): 149.3 (C2), 125.1 (C3), 147.0 (C4), 129.6 (C4a), 66.2 (C5), 70.3 (C6), 137.7 (C7), 128.6 (C8), 152.1 (C8a), 17.7 (CH3).
The product was isolated by flash chromatography of preparative experiments and identified as 6-methylquinolin-3-ol (4h3) by 1H and 13C NMR.
6-Methylquinolin-3-ol (4h3). δ H (500 MHz, (CD3)2CO): 8.59 (1H, d, 4J2–4 = 2.5, 2-H), 6.09 (1H, d, 4J4–2 = 2.5, 4-H), 7.54 (1H, br s, 5-H), 7.38 (1H, dd, J7–8 = 8.6, 4J7–5 = 1.7, 7-H), 7.87 (1H, d, J8–7 = 8.6, 8-H), 2.51 (3H, s, CH3); δC (125 MHz, (CD3)2CO): 144.3 (C2), 152.6 (C3), 116.6 (C4), 138.1 (C4a), 127.0 (C5), 131.1 (C6), 129.8 (C7), 130.5 (C8), 143.8 (C8a), 22.4 (CH3).
Flash chromatography afforded two products which were identified by 1H and 13C NMR as cis-8-methyl-5,6-dihydroquinoline-5,6-diol (5d1) and quinolin-8-ylmethanol (5e). They were also obtained by RP-chromatography over a prepacked C18 column and elution with H2O–CH3CN in the appropriate ratios.
cis-8-Methyl-5,6-dihydroquinoline-5,6-diol (5d1). [αD] + 222 (c 0.035, MeOH); δH (250 MHz, (CD3)2CO): 8.42 (1H, br d, J2–3 = 4.4, 2-H), 7.21 (1H, dd, J3–4 = 7.5, J2–3 = 4.8, 3-H), 7.84 (1H, d, J4–3 = 7.4, 4-H), 4.67 (1H, d, J5–6 = 4.7, 5-H), 4.23 (1H, t, J = 4.8, 6-H), 6.17 (1H, d, J7–6 = 3.6, 7-H), 2.11 (3H, br s, CH3); δC (125 MHz, (CD3)2CO): 148.6 (C2), 123.1 (C3), 134.5 (C4), 134.2 (C4a), 71.0 (C5), 67.0 (C6), 130.1 (C7), 137.5 (C8), 153.7(C8a), 18.3 (CH3).
(4-Bromo)phenylboronate derivative (5d1B). 5d1 (85 mg, 0.48 mmol) and 4-bromophenyl boronic acid (105 mg, 1.1 equiv.) were reacted in CH2Cl2/(CH3)2CO 1/1 (5 cm3) at room temperature during 1 h. The product formed was purified by flash chromatography with CH2Cl2 as eluent. δH (250 MHz, (CD3)2CO): 8.56 (1H, dd, J2–3 = 4.8, 4J2–4 = 1.6, 2-H), 7.34 (1H, dd, J3–4 = 7.7, J3–2 = 4.8, 3-H), 7.93 (1H, dd, J4–3 = 7.7, 4J2–4 = 1.5, 4-H), 5.49 (1H, d, J5–6 = 9.0, 5-H), 5.27 (1H, dm, J6–5 = 9.0, 6-H), 6.21 (1H, dt, J7–6 = 3.8, 4J7-CH3 = 1.6, 7-H), 2.16 (3H, t, JCH3-7 = 1.6 CH3), 7.51–7.62 (4H, m, Ar–H). The crystals grown by slow evaporation from the CH2Cl2 solution were not suitable for X-ray analysis.
Quinolin-8-ylmethanol (5e). δ H (500 MHz, (CD3)2CO): 8.89 (1H, dd, J2–3 = 4.3, 4J2–4 = 1.8, 2-H), 7.53 (1H, dd, J3–4 = 8.2, J3–2 = 4.2, 3-H), 8.34 (1H, dd, J4–3 = 8.1, 4J2–4 = 1.6, 4-H), 7.82 (1H, d, J5–6 = 7.7, 5-H), 7.58 (1H, dd, J6–7 = 8.1, J6–5 = 7.7, 6-H), 7.85 (1H, d, J7–6 = 8.1, 7-H), 5.25 (2H, s, CH2); δC (125 MHz, (CD3)2CO): 150.2 (C2), 122.1 (C3), 137.3 (C4), 129.1 (C4a), 127.3 (C5), 127.2 (C6), 127.6 (C7), 140.8 (C8), 147.1(C8a), 62.5 (CH2).
SP-flash chromatography isoquinolin-5-ol (6h5), with a small amount of an other phenol (6h4) were eluted with CH2Cl2; cis-7,8-dihydroisoquinoline-7,8-diol (6d2) and cis-5,6-dihydroisoquinoline-5,6-diol (6d1) were eluted with CH2Cl2/CH3OH (0→10%). 6h5, 6d1, and 6d2 were identified by comparing their 1H NMR spectra to published data.9 The structure of 6h4 was deduced from 1H and 13C NMR analysis and assigned to isoquin-4-ol. The yield of 6h5, 6d2, 6h4, and 6d1 for this biotransformation were evaluated to 55, 6, 5, and 4%, respectively by NMR analysis of the crude organic extract.
Isoquin-4-ol (6h4). δH (500 MHz, (CD3)2CO): 8.82 (1H, s, 1-H), 8.12 (1H, s, 3-H), 8.21 (1H, d, J5–6 = 8.3, 5-H), 7.73 (1H, t, J = 7.5, 6-H), 7.66 (1H, t, J = 7.5, 7-H), 8.03 (1H, d, J8–7 = 8.3, 8-H); δC (125 MHz, (CD3)2CO): 144.5 (C1), 127.9 (C3), 149.0 (C4), 127.2 (C4a),121.7 (C5), 129.7 (C6), 128.3 (C7), 127.6 (C8), 130.4 (C8a).
As our attempts to obtain a pure sample of cis-7,8-dihydroisoquinoline-7,8-diol (6d2) were not successful, the sample used to determine the optical activity of 6d2 was a mixture of 6h5, 6d1, and 6d2, in the proportions of 8, 21, and 71% respectively determined by 1H NMR. The value of the optical activity of the sample was [α]D + 97.5 (c 0.56, MeOH); using the value of + 163 for the optical activity of 6d1,9 the estimated value for 6d2 is then + 134.
RP-flash chromatography afforded two products which were identified by 1H and 13C NMR analysis as cis-1-methyl-7,8-dihydroisoquinoline-7,8-diol (7d2) and isoquinolin-1-ylmethanol (7e) according to Weitgenant et al.20 As shown by NMR analysis of the crude organic extract, only these two products are formed in the biotransformation of 1-methyl isoquinoline.
cis-1-Methyl-7,8-dihydroisoquinoline-7,8-diol (7d2). [α]D + 25 (c 0.35, MeOH); δH (500 MHz, (CD3)2CO): 8.31 (1H, d, J3–4 = 5.0, 3-H), 6.92 (1H, d, J4–3 = 5.0, 4-H), 6.38 (1H, dd, J5–6 = 9.8, J = 2.7, 5-H), 6.08 (1H, dt, J6–5 = 9.8, J = 1.6, 6-H), 4.51 (1H, m, 7-H), 4.79 (1H, dd, J8–7 = 5.2,J = 1.3, 8-H), 2.57 (3H, s, CH3); δC (125 MHz, (CD3)2CO): 158.1 (C1), 149.9 (C3), 119.5 (C4), 140.2 (C4a), 125.4 (C5), 138.2 (C6), 70.6 (C7), 65.9(C8), 128.4 (C8a), 21.1 (CH3).
(4-Bromo)phenylboronate derivative of cis-1-methyl-7,8-dihydroisoquinoline-7,8-diol (7d2B). 7d2 (85 mg, 0.48 mmol) and 4-bromophenyl boronic acid (105 mg, 1.1 equiv.) were reacted in CH2Cl2 (5 cm3) at room temperature during 1 h. The product formed was purified by flash chromatography with CH2Cl2 as eluent; δH (250 MHz, (CD3)2CO): 8.42 (1H, d, J3–4 = 4.9, 3-H), 7.02 (1H, d, J4–3 = 4.9, 4-H)), 6.56 (1H, dd, J5–6 = 9.9, 4J5–7 = 1.5, 5-H), 6.21 (1H, dd, J6–5 = 10.0, J6–7 = 3.0, 6-H), 5.60 (1H, dm, J7–8 = 9.6, 7-H), 5.92 (1H, d, J8–7 = 9.6, 8-H), 2.71(3H, s, CH3), 7.56–7.76 (4H, m, Ar–H). Crystals suitable for X-ray analysis were grown by slow evaporation from the CH2Cl2 solution. They were obtained as a mass of thin needles 10 micron thick. After dispersion in paraffin oil, one needle was selected, fished out using a standard cryo-loop and frozen in liquid nitrogen. Diffraction data were recorded at ESRF synchrotron (Grenoble), beam line BM 30A (FIP). Wavelength was set to 0.914 Å so as to get a significant anomalous signal from the bromine K-edge. Crystal data for 7d2B: monoclinic, P21 with parameters: a = 12.683(2); b = 4.267(2); c = 14.339(2) Å; β = 107.93(5)° and Z = 2. The structure was refined to R = 8.5% (1629 observed Fo) and R = 8.6% (all 1655 Fo data).
RP-flash chromatography afforded one product which was identified by 1H and 13C NMR as cis-3-methyl-7,8-dihydroisoquinoline-7,8-diol (8d2).
cis-3-Methyl-7,8-dihydroisoquinoline-7,8-diol (8d2). [α]D + 192 (c 3.4, MeOH); δH (500 MHz, (CD3)2CO): 8.43 (1H, s, 1-H), 6.84 (1H, s, 4-H), 6.38 (1H, dd, J5–6 = 9.8, J = 2.7, 5-H), 6.08 (1H, dt, J6–5 = 9.8, J = 1.6, 6-H), 4.37 (1H, m, 7-H), 4.65 (1H, d, J7–8 = 4.9, 8-H), 2.44 (3H, s, CH3); δC (125 MHz, (CD3)2CO): 148.8 (C1), 159.3 (C3), 120.3 (C4), 140.4 (C4a), 126.6 (C5), 136.6 (C6), 68.6 (C7), 68.5(C8), 129.0 (C8a), 24.2 (CH3).
A monohydroxylated metabolite was isolated by SP-flash chromatography and identified by 1H and 13C NMR as 3-methylisoquinolin-5-ol (8h5).
3-Methyl-isoquinolin-5-ol (8h5). δ H (500 MHz, (CD3)2CO): 9.10 (1H, s, 1-H), 7.84 (1H, s, 4-H), 7.09 (1H, d, J7–8 = 7.6, 6-H), 7.37 (1H, t, J = 7.9 7-H), 7.51 (1H, d, J8–7 = 8.1, 8-H), 2.64 (3H, s, CH3); δC (125 MHz, (CD3)2CO): 152.2 (C1), 151.3 (C3), 113.8 (C4), 129.2 and 129.1 (C4a, C8a), 152.9 (C5), 112.8 (C6), 127.7 (C7), 119.1 (C8), 24.14 (CH3).
(4-Bromo)phenylboronate derivative of cis-3-methyl-7,8-dihydroisoquinoline-7,8-diol (8d2B). 8d2 (85 mg, 0.48 mmol) and 4-bromophenyl boronic acid (105 mg, 1.1 equiv.) were reacted in CH2Cl2 (5 cm3) at room temperature during 1 h. The product formed was purified by flash chromatography with CH2Cl2 as eluent. δH (250 MHz, (CD3)2CO): 8.59 (1H, s, 1-H), 7.04 (1H, s, 4-H), 6.56 (1H, d, J5–6 = 9.9, 5-H), 6.21 (1H, dd, J6–5 = 9.9, J6–7 = 3.2, 6-H), 5.55 (1H, ddd, J7–8 = 9.1, J7–6 = 3.2, 4J7–5 = 1.4, 7-H), 5.78 (1H, d, J8–7 = 9.0, 8-H), 2.48 (3H, s, CH3), 7.56–7.73 (4H, m, Ar-H). Crystals grown by slow evaporation from the CH2Cl2–Et2O were not suitable for X-ray analysis.
:9 ratio, with a biotransformation which reached only ≈ 20%. There is no evidence of the presence of a dihydrodiol in the crude extract. Its purification by flash chromatography with CH2Cl2 as eluent gave only a monohydroxylated compound identified as phthalazin-5-ol (9h5) by NMR analysis.
Phthalazin-5-ol (9h5). δ H (500 MHz, (CD3)2CO): 9.50 (1H, d, 5J1–4 = 1.4, 1-H), 9.72 (1H, s, 4-H), 7.35 (1H, dd, J6–7 = 7.5, J = 1.0, 6-H), 7.83 (1H, dd, J7–8 = 7.9, J7–6 = 7.5, 7-H), 7.57 (1H, d, J8–7 = 7.9, 8-H); δC (125 MHz, (CD3)2CO): 151.8 (C1), 147.1 (C4), 118.4 (C4a), 154.7 (C5), 116.7 (C6), 135.2 (C7), 117.7 (C8), 128.6 (C8a).
:CH3OH 100:0 to 90:10) afforded two products, identified by 1H and 13C NMR analysis as quinazolin-8-ol (11h8) and the previously described cis-5,6-dihydroquinoxaline-5,6-diol (11d1).9 This dihydrodiol is opticaly active: [α]D + 197 (c 0.35, MeOH).
(4-Bromo)phenylboronate derivative of cis-5,6-dihydroquinazoline-5,6-diol (11d1B). 11d1 (15 mg, 0.09 mmol) and 4-bromophenyl boronic acid (20 mg, 1.1 equiv.) were reacted in CH2Cl2/(CH3)2CO 1/1 (2 cm3) at room temperature during 1 h. The product formed was purified by flash chromatography with CH2Cl2 as eluent. δH (250 MHz, (CD3)2CO): 8.90 (1H, s, 2-H), 9.08 (1H, s, 4-H), 5.93 (1H, d, J5–6 = 9.4, 5-H), 5.65 (1H, dd, J6–5 = 9.4, 4J6–8 = 2.2, 6-H), 6.50–6.70 (2H, m, 7-H and 8-H), 7.50–7.80 (4H, m, Ar–H). The crystals grown by slow evaporation from the CH2Cl2 solution were not suitable for X-ray analysis.
Quinazolin-8-ol (11h8). δ H (500 MHz, (CD3)2CO): 9.22 (1H, s, 2-H), 9.50 (1H, s, 4-H), 7.58 (1H, dd, J5–6 = 8.3, J5–7 = 1.4, 5-H), 7.62 (1H, dd, J6–5 = 7.9, J6–7 = 7.4, 6-H), 7.37 (1H, dd, J7–6 = 7.4 and J7–5 = 1.4, 7-H); δC (125 MHz, (CD3)2CO): 154.5 (C2), 161.2 (C4), 126.6 (C4a), 118.3 (C5), 129.9 (C6), 116.1 (C7), 153.4 (C8), 141.1 (C8a).
Acknowledgements
We are indebted to Pr Gibson (Iowa state University) for the strain E.coli JM109(DE3)(pDTG141). We would like to thank Dr R. Azerad and Dr D. Mansuy (UMR 8601, Université Paris Descartes, Paris) who made valuable suggestions on reading the draft of this manuscript, and the staff of the BM30A beamline at the European Synchrotron Facility, Grenoble for beamtime allocation.References
- T. Hudlicky, D. Gonzalez and D. T. Gibson, Aldrichimica Acta, 1999, 32, 35–62 CAS.
- M. A. Endoma, V. P. Bui, J. Hansen and T. Hudlicky, Org. Process Res. Dev., 2002, 6, 525–532 CrossRef CAS.
- D. R. Boyd and T. D. H. Bugg, Org. Biomol. Chem., 2006, 4, 181–192 CAS.
- L. Modyanova and R. Azerad, Tetrahedron Lett., 2000, 41, 3865–3869 CrossRef CAS.
- D. R. Boyd, N. D. Sharma, L. V. Modyanova, J. G. Carroll, J. F. Malone, C. C. R. Allen, J. T. G. Hamilton, D. T. Gibson, R. E. Parales and H. Dalton, Can. J. Chem., 2002, 80, 589–600 CrossRef CAS.
- C. Chopard, R. Azerad and T. Prangé, J. Mol. Catal. B: Enzym., 2008, 50, 53–60 CrossRef CAS.
- S. M. Resnick, D. S. Torok and D. T. Gibson, FEMS Microbiol. Lett., 1993, 113, 297–302 CrossRef CAS.
- D. R. Boyd, R. A. S. McMordie, H. P. Porter, H. Dalton, R. O. Jenkins and O. W. Howarth, J. Chem. Soc., Chem. Commun., 1987, 1722–1724 RSC.
- D. R. Boyd, N. D. Sharma, M. R. Dorrity, M. V. Hand, R. A. S. Mcmordie, J. F. Malone, H. P. Porter, H. Dalton, J. Chima and G. N. Sheldrake, J. Chem. Soc., Perkin Trans. 1, 1993, 1065–1071 RSC.
- D. R. Boyd, N. D. Sharma, J. G. Carroll, J. F. Malone, D. G. Mackerracher and C. C. R. Allen, Chem. Commun., 1998, 683–684 RSC.
- D. R. Boyd, N. D. Sharma, G. P. Coen, F. Hempenstall, V. Ljubez, J. F. Malone, C. C. R. Allen and J. T. G. Hamilton, Org. Biomol. Chem., 2008, 6, 3957–3966 CAS.
- E. A. Bleeker, S. Wiegman, P. de Voogt, M. Kraak, H. A. Leslie, E. de Haas and W. Admiraal, Rev. Environ. Contam. Toxicol., 2002, 173, 39–83 Search PubMed.
- D. L. Tullis and S. Banerjee, Cancer Lett., 1984, 23, 241–244 CrossRef CAS.
- E. J. LaVoie, S. Dolan, P. Little, C. X. Wang, S. Sugie and A. Rivenson, Food Chem. Toxicol., 1988, 26, 625–629 CrossRef CAS.
- M. Dua, A. Singh, N. Sethunathan and A. K. Johri, Appl. Microbiol. Biotechnol., 2002, 59, 143–152 CrossRef CAS.
- J. Aislabie, D. J. Saul and J. M. Foght, Extremophiles, 2006, 10, 171–179 CrossRef CAS.
- S. Fetzner, B. Tshisuaka, F. Lingens, R. Kappl and J. Huttermann, Angew. Chem., Int. Ed., 1998, 37, 577–597 CrossRef.
- H. D. Flack, Acta Crystallogr., 1983, A39, 876–881 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef CAS.
- J. A. Weitgenant, J. D. Mortison and P. Helquist, Org. Lett., 2005, 7, 3609–3612 CrossRef CAS.
- S. M. Resnick, K. Lee and D. T. Gibson, J. Ind. Microbiol., 1996, 17, 438–457 CrossRef CAS.
- G. Bestetti, D. Bianchi, A. Bosetti, P. DiGennaro, E. Galli, B. Leoni, F. Pelizzoni and G. Sello, Appl. Microbiol. Biotechnol., 1995, 44, 306–313 CrossRef CAS.
- D. R. Boyd, N. D. Sharma, I. N. Brannigan, S. A. Haughey, J. F. Malone, D. A. Clarke and H. Dalton, Chem. Commun., 1996, 2361–2362 RSC.
- A. Karlsson, H. Eklund, S. Ramaswamy, J. V. Parales, R. E. Parales and D. T. Gibson, J. Inorg. Biochem., 2003, 96, 164 CrossRef.
- D. M. Jerina, H. Selander, H. Yagi, M. C. Wells, J. F. Davey, V. Mahadevan and D. T. Gibson, J. Am. Chem. Soc., 1976, 98, 5988–5996 CrossRef CAS.
- A. Karlsson, J. V. Parales, R. E. Parales, D. T. Gibson, H. Eklund and S. Ramaswamy, Science, 2003, 299, 1039–1042 CrossRef CAS.
- R. E. Parales, S. M. Resnick, C. L. Yu, D. R. Boyd, N. D. Sharma and D. T. Gibson, J. Bacteriol., 2000, 182, 5495–5504 CrossRef CAS.
- D. R. Boyd, N. D. Sharma, L. Sbircea, D. Murphy, T. Belhocine, J. F. Malone, S. L. James, C. C. R. Allen and J. T. G. Hamilton, Chem. Commun., 2008, 5535–5537 RSC.
- M. Kwit, J. Gawronski, D. R. Boyd, N. D. Sharma, M. Kaik, R. A. More O'Ferrall and J. S. Kudavalli, Chem.–Eur. J., 2008, 14, 11500–11511 CrossRef CAS.
Footnote |
| † CCDC reference numbers 815425–815426. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ra00706h |
| This journal is © The Royal Society of Chemistry 2012 |