Highly efficient green-emitting electrophosphorescent hyperbranched polymers using a bipolar carbazole-3,6-diyl-co-2,8-octyldibenzothiophene-S,S-dioxide-3,7-diyl unit as the branch
Jie
Liu
,
Lei
Yu
,
Chengmei
Zhong
,
Ruifeng
He
,
Wei
Yang
*,
Hongbin
Wu
* and
Yong
Cao
State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou, 510640, China. E-mail: pswyang@scut.edu.cn; Fax: +86-020-87110606; Tel: +86-020-87114346-17
First published on 21st November 2011
Abstract
Green-emitting hyperbranched polymers with a bipolar 3,6-carbazole-co-2,8-dioctyl-dibenzothiophene-S,S-dioxide-3,7-diyl [Cz-co-DOSO] as the branch and fac-tris(2-phenylpyridine)iridium[Ir(ppy)3] as the core were synthesized by Suzuki polycondensation. The Cz-co-DOSO branch with a triplet energy level above 2.5 eV can effectively prevent energy feedback from the phosphorescent core. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) levels of the copolymers reduce dramatically by over 0.6 and 0.8 eV with increasing DOSO content up to 45.5 mol% in the polymers, respectively. A maximum luminous efficiency of 50.5 cd A−1 and a maximum external quantum efficiency of 15.3% were obtained for PCzDOSO40Ir3 based on a single layer device of ITO/PEDOT:PSS/polymer/CsF/Al, which were improved by over 13 times compared with PCzIr3. The hole- and electron-only devices show that the hole and electron flux are well balanced, which demonstrates that PCzDOSO40Ir3 is a bipolar polymer with a balanced charge carrier transport.
Introduction
In the past years, significant effort has been focused on developing highly efficient phosphorescent materials,1 because phosphorescent dyes can capture both singlet and triplet excitons, with the internal quantum efficiency near 100%.2 Among them, polymeric materials are of special interest due to their facile processability from solution by spin-coating or ink-jet printing technologies.3 Phosphorescent emitters were often physically or chemically embedded within an appropriate polymeric matrix to reduce concentration quenching and triplet–triplet annihilation.4–5 Irrespective of the kind of methods used, the host should ideally fulfil some basic requirements,6 such as higher triplet energy level than phosphorescent emitter to prevent energy feedback from the guest to the host, and good overlap for the host emission with the guest absorption to ensure effective charge and energy transfer.It is well known that blended systems inherently involve the risk of undesired phase separation, aggregation or crystallization, which can harm the device performance. Therefore, phosphorescent polymers with complexes in the molecular chain have received increasing attention. A promising strategy to balance charge transport is incorporating both electron-donating and electron-withdrawing units into a polymer backbone. However, the triplet energy level of the host segment might be reduced.7a,7b To date, developing phosphorescent polymers with bipolar transporting properties still remains a challenge.
Carbazole-based polymers are potential host candidates for phosphorescent emitters due to their high triplet energy level and hole transport ability.7 For example, Park et al. reported wide-gap non-conjugated carbazole-based polymers exploiting a FIrpic unit as the pendant with a luminous efficiency (LE) of 2.23 cd A−1.8 Zhen et al. synthesized 3,6-carbazole-based green light-emitting conjugated polymers with a LE of 4.4 cd A−1.9 Dijken et al. reported a green light-emitting diode by doping a green triplet emitter in the carbazole-oxadiazole host copolymer with a LE of 23 cd A−1.7a
In parallel with linear polymers, hyperbranched polymers have been proposed as an alternative approach to embed a phosphorescent emitter within polymeric systems due to facile tunable properties through modification of the core, branch or surface groups.10 A more important point is that the phosphorescent emitter could be separated effectively. Guan et al. designed green-emitting hyperbranched phosphorescent polymers using carbazole-co-pyridine as the host segment branch.11 Although high efficiencies were achieved, 2-(4-bisphenylyl)-5-(4-tert-butylphenyl)-1,3,4-oxadiazole (PBD) was still blended as an electron transport material to facilitate electron transport in a single active layer configuration like ITO/PEDOT/polymer + PBD (30 wt%)/Ba/Al.
Dibenzothiophene-S,S-dioxide is an electron deficient moiety with high fluorescence efficiency, electron affinity energy and electron transport properties.12 Recently, a 2,8-dialkyl-substituted dibenzothiophene-S,S-dioxide-3,7-diyl unit was incorporated into polyfluorene backbone and raised the triplet energy level of the polymer to 2.46 eV.13 These results indicate that the alkylated dibenzothiophene-S,S-dioxide unit is a potential candidate to modify host materials. In this contribution, green-emitting hyperbranched polymers utilizing a bipolar carbazole-3,6-diyl-co-2,8-dioctyldibenzothiophene-S,S-dioxide-3,7-diyl as the branch and an Ir(ppy)3 as the core were synthesized and studied.
Results and discussion
Synthesis and characterization
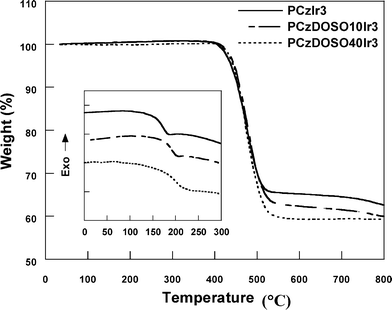
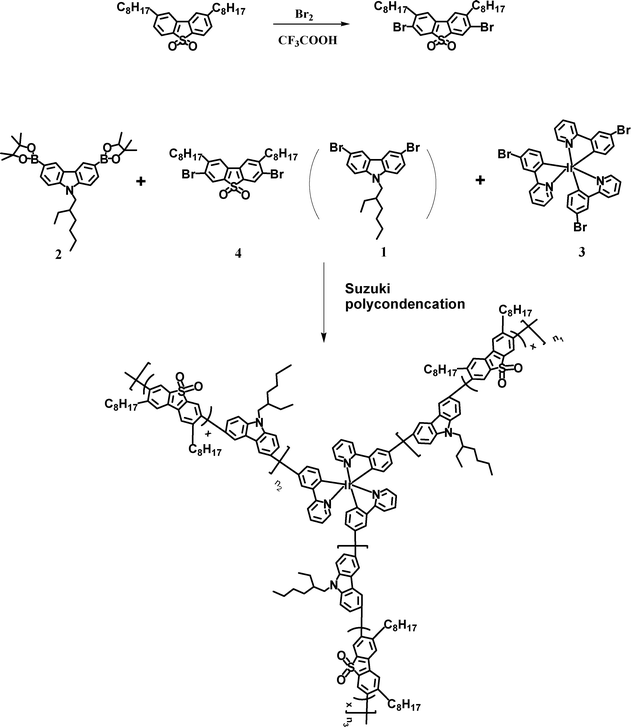
The synthesis of 3,7-dibromo-2,8-dioctyldibenzothiophene-S,S-dioxide even reported used concentrated H2SO4 and N-bromosuccinimide as the bromination system.13 But the process has a low conversion with a yield under 40%, due to the poor solubility of 2,8-dioctyldibenzothiophene-S,S-dioxide in H2SO4 solution. Here, the bromination of 2,8-dioctyldibenzothiophene-S,S-dioxide was processed in trifluoroacetic acid and bromine solution, and obtained a yield of over 70%. The synthetic route of the polymers is depicted in Scheme 1. A series of hyperbranched copolymers with carbazole-3,6-diyl-co-2,8-dioctyldibenzothiophene-S,S-dioxide-3,7-diyl as the branch and Ir(ppy)3 as the core were synthesized by Suzuki polycondensation. The feed ratio of the iridium complex as well as the DOSO moiety was 1, 2 or 3, and 45.5, 40, 30, 20, 10, or 0 mol%, and the corresponding polymers were named as PCzDOSO45Ir1, PCzDOSO45Ir2, PCzDOSO45Ir3, PCzDOSO40Ir3, PCzDOSO30Ir3, PCzDOSO20Ir3, PCzDOSO10Ir3 and PCzIr3, respectively. It is found that the actual ratios of Ir-complex in the polymers are substantially lower than the feed ratios (Table 1). The number-average molecular weights (Mns) of the polymers range from 5800 to 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 with the polydispersity indices (PDI) in the range 1.36∼2.60. It is noted that the Mns increase with increasing the content of DOSO from 0 to 45.5 mol% in copolymers, and is suggested that the 3,7-dibromo-2,8-dioctyldibenzothiophene-S,S-dioxide monomer has a high reactive activity in Suzuki polycondensation.11Fig. 1 shows the TGA and DSC curves of the polymers. Compared with the glass transition temperature (Tg) of PCzIr3 (175 °C), the Tg of PCzDOSO40Ir3 increases to 205 °C, and Tgs increase with increasing the content of the DOSO unit at the same content of iridium complex in the polymers. The decomposition temperatures (Tds, corresponding to 5 wt% weight loss) are all around 430 °C, as shown in Table 1. Thermal properties could be effectively enhanced by the introduction of a DOSO unit into the polymers.
500 with the polydispersity indices (PDI) in the range 1.36∼2.60. It is noted that the Mns increase with increasing the content of DOSO from 0 to 45.5 mol% in copolymers, and is suggested that the 3,7-dibromo-2,8-dioctyldibenzothiophene-S,S-dioxide monomer has a high reactive activity in Suzuki polycondensation.11Fig. 1 shows the TGA and DSC curves of the polymers. Compared with the glass transition temperature (Tg) of PCzIr3 (175 °C), the Tg of PCzDOSO40Ir3 increases to 205 °C, and Tgs increase with increasing the content of the DOSO unit at the same content of iridium complex in the polymers. The decomposition temperatures (Tds, corresponding to 5 wt% weight loss) are all around 430 °C, as shown in Table 1. Thermal properties could be effectively enhanced by the introduction of a DOSO unit into the polymers.
 | ||
| Scheme 1 Synthetic route of the monomer and polymers. | ||
| Polymer | M n (×103) | PDI | Ir-complex (mol%) in | T d a/Tg/°C | E g b/eV | E HOMO c/eV | E LUMO/eV | |
|---|---|---|---|---|---|---|---|---|
| Feed ratio | Polymer | |||||||
| a Corresponding to 5 wt.% loss. b Calculated from the absorption spectra threshold. c Estimated from the HOMO levels and the optical band gaps. | ||||||||
| PCzDOSO45Ir1 | 9.8 | 2.60 | 1 | 0.69 | 435/175 | 3.07 | −5.86 | −2.79 |
| PCzDOSO45Ir2 | 11.7 | 2.60 | 2 | 1.68 | 435/173 | 3.07 | −5.84 | −2.77 |
| PCzDOSO45Ir3 | 36.5 | 1.38 | 3 | 2.80 | 429/206 | 3.07 | −5.85 | −2.78 |
| PCzDOSO40Ir3 | 28.0 | 1.42 | 3 | 2.49 | 444/205 | 3.07 | −5.78 | −2.71 |
| PCzDOSO30Ir3 | 12.8 | 2.31 | 3 | 2.75 | 430/202 | 3.07 | −5.66 | −2.59 |
| PCzDOSO20Ir3 | 11.4 | 2.50 | 3 | 2.17 | 446/193 | 3.13 | −5.30 | −2.17 |
| PCzDOSO10Ir3 | 8.7 | 1.99 | 3 | 1.88 | 447/185 | 3.21 | −5.27 | −2.06 |
| PCzIr3 | 5.8 | 1.36 | 3 | 2.21 | 439/175 | 3.29 | −5.21 | −1.92 |
Electrochemical properties
The electrochemical behaviors of the resulting polymers were examined by cyclic voltammetry (CV). As shown in Fig. 2, the oxidation potentials (Eoxs) are enhanced when the content of DOSO increases from 0 to 45.5 mol% in the copolymers. The reference polymer PCzIr3 exhibits an oxidation potential at 0.81 V, while the polymers PCzDOSO45Ir show an almost identical oxidation potential at about 1.45 V, which is shifted positively by about 0.64 V. The HOMO levels determined from the onsets of the oxidation potentials are −5.21 eV and −5.85 eV for PCzIr3 and PCzDOSO45Ir3, respectively, by assuming the energy level of Fc/Fc+ to be 4.8 eV below the vacuum level.14 Unfortunately, we were unable to record n-doping processes after many attempts. In order to get the LUMO levels of these polymers, the LUMO levels calculated from the HOMO levels and corresponding optical bandgaps. As shown in Table 1, it is found that the LUMO levels of PCzDOSO45Ir3 reduce significantly by over 0.8 eV with respect to that of PCzIr3, indicating that the incorporation of the DOSO moiety into the backbone could effectively improve the electron injection of the polymers.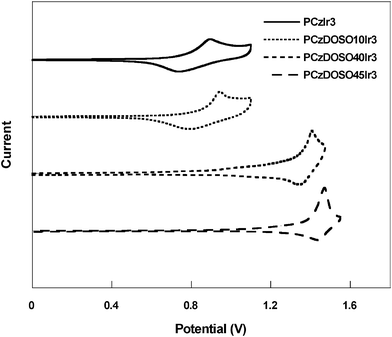 | ||
| Fig. 2 Cyclic voltammetry curves of the polymers. | ||
Photophysical properties
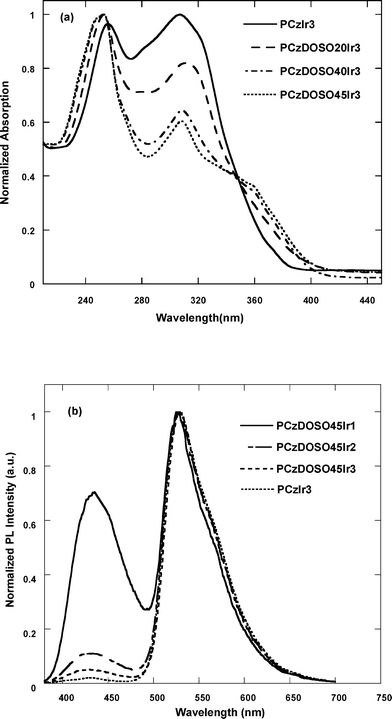
The phosphorescent spectrum of poly(N-(2-ethylhexyl)-carbazole-3,6-diyl-alt-2,8-dioctyldibenzothiophene-S,S-dioxide-3,7-diyl) [P(Cz-alt-DOSO)] at 77 K shows the onset phosphorescence at 493 nm (Fig. 3), corresponding to the triplet energy level of 2.5 eV. It is higher than the triplet energy level of Ir(ppy)3 (ET ≈ 2.4 eV),15 indicating that the Cz-alt-DOSO segment can efficiently prevent the energy feedback from the core. Additionally, the triplet energy level of poly(3,6-carbazole) is 2.6 eV,7a which indicates that the Cz-co-DOSO branch could be suitable for green-emitting phosphorescent polymers. Fig. 4a shows the UV-Vis absorption spectra of the polymers. The absorption peaks at 256 and 306 nm are from the 3,6-carbazole unit,16 and at about the 360 nm peak corresponds to the DOSO unit.13a It is shown that the absorption spectra peaks at 306 nm decline gradually with increasing the content of DOSO. The absorption band of the Ir-complex was not detected due to the comparatively low content in these copolymers. PL spectra of the polymers are depicted in Fig. 4b. All polymers show two bands at around 435 and 528 nm, which are assigned to the branches and complex emission, respectively. As the content of complex increases, the emission intensity around 435 nm from the branch segment decreases. It is found that PCzDOSO45Ir1 shows two comparable emission bands, while PCzDOSO45Ir3 exhibits dominant green emission.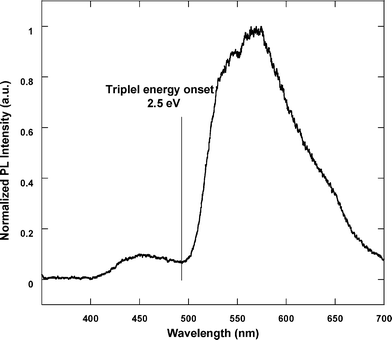 | ||
| Fig. 3 Phosphorescent spectrum of PCz-alt-DOSO at 77 K. | ||
Electroluminescent properties
The devices were fabricated with the structure ITO/PEDOT:PSS/polymer/CsF/Al. Fig. 5 shows EL spectra of the polymers. EL spectra from the polymers with a higher content of Ir-complex only exhibit green emission, indicating that the energy transfer from the host site to guest phosphor core is very efficient when the Ir-complex content in the polymers is over 1 mol%.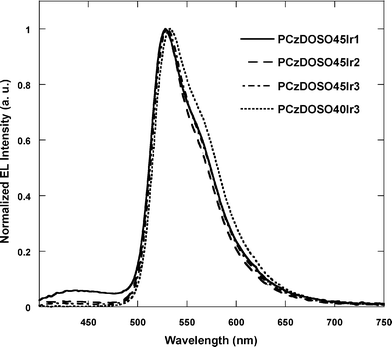 | ||
| Fig. 5 EL spectra of the polymers. | ||
The detailed EL performances are summarized in Table 2. With the increase of Ir-complex in the polymers from 1 mol% to 3 mol%, the luminous efficiencies (LE) were improved from 7.8 cd A−1, 14.9 cd A−1 to 20.7 cd A−1, respectively. After establishing that 3 mol% was the optimized concentration for the core and upon fixing the Ir-complex content, the contents of DOSO were changed from 0 mol% to 45.5 mol%. As can be clearly seen in Fig. 6a, the operation voltages corresponding to a given current density (i.e., 50 mA cm−2) increase steadily upon the increase of DOSO content from 0 mol% to 40 mol%. This can be understood since charge transport in PCzIr3 is dominated by holes (characterized by unbalanced charge transport properties with a higher hole mobility than the electron), while the incorporation and increased content of the electron deficient unit, DOSO, result in deeper LUMO/HOMO levels (Fig. 6c) and facilitate electron transport. In other words, with the increase of an electron deficient unit, both hole flux (due to higher injection barrier) and charge mobility decrease while in electron flux and its mobility increase. As expected, optimized device performances were obtained in higher DOSO content (PCzDOSO40Ir3) based devices, with a maximal LE of 50.5 cd A−1, while other polymers with the DOSO content of 0–20 mol% only show moderated LEs in the range 3.8–13.4 cd A−1. To the best of our knowledge, the high LE over 50 cd A−1 in this study is one of the highest efficiencies of green PLEDs among electrophosphorescent polymers.9,11,17
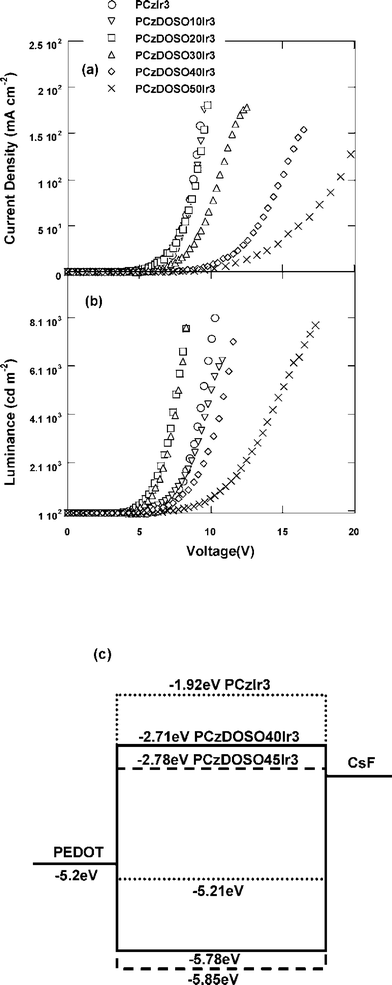 | ||
| Fig. 6 Current density–voltage (J–V) (a), luminance–voltage (L–V) (b) curves and energy level diagrams of the devices (c). | ||
| Polymer | V on a/V | Lmax/cd m−2 | LEmax/cd A−1 | EQEmax(%) | J = 100 mA cm−2 | CIE b (x, y) | ||
|---|---|---|---|---|---|---|---|---|
| LE/cd A−1 | EQE (%) | L cd−1 m−2 | ||||||
| a Corresponding to 1 cd m−2. b Measured at 17.6 mA cm−2. The device structure of ITO/PEDOT:PSS/polymer/ CsF/Al. | ||||||||
| PCzDOSO45Ir1 | 5.4 | 3761 | 7.8 | 3.0 | 3.3 | 1.3 | 3318 | 0.33, 0.62 |
| PCzDOSO45Ir2 | 5.5 | 12601 | 14.9 | 5.8 | 8.9 | 3.5 | 8840 | 0.34, 0.62 |
| PCzDOSO45Ir3 | 4.4 | 9918 | 20.7 | 6.5 | 8.8 | 2.8 | 9014 | 0.37, 0.60 |
| PCzDOSO40Ir3 | 3.5 | 22581 | 50.5 | 15.3 | 25.0 | 6.8 | 21784 | 0.37, 0.61 |
| PCzDOSO30Ir3 | 3.2 | 22147 | 45.7 | 14.2 | 21.6 | 6.9 | 22124 | 0.38, 0.60 |
| PCzDOSO20Ir3 | 3.5 | 13126 | 13.4 | 3.9 | 9.1 | 2.5 | 8349 | 0.38, 0.60 |
| PCzDOSO10Ir3 | 3.5 | 7307 | 6.7 | 2.0 | 4.3 | 1.3 | 5096 | 0.37, 0.60 |
| PCzIr3 | 3.8 | 12001 | 3.8 | 1.1 | 3.7 | 1.1 | 3695 | 0.38, 0.60 |
To further clarify the bipolar charge transport properties of the polymer, single charge carrier devices from PCzDOSO40Ir3 were fabricated and their current density–voltage characteristics (J–V) were studied. As shown in Fig. 7, both of the hole- and electron-only devices exhibited nearly identical current density at the entire applied voltage range (0–8 V), thus balanced hole and electron mobilities for this polymer are anticipated, revealing bipolar charge-transport property.
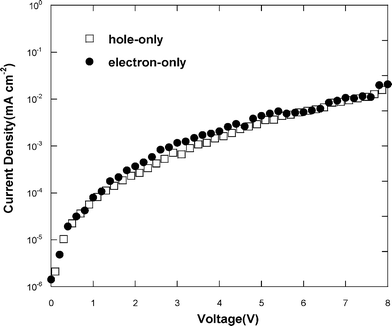 | ||
| Fig. 7 Current density–voltage (J–V) characteristics of the hole- and electron-only devices based on PCzDOSO40Ir3. | ||
Fig. 8 displays the luminous efficiency–current density–luminance (LE–J–L) characteristics of the resulting copolymers. The luminance increases and LE declines slowly with the increase of current density, which could be assigned that the phosphorescent core could be separated efficiently by the hyperbranched framework to reduce the concentration quenching of the cores.
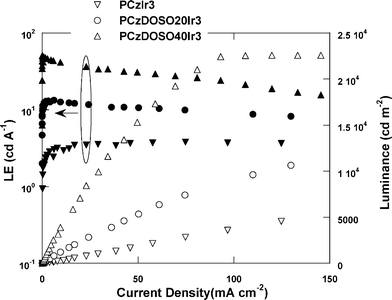 | ||
| Fig. 8 Luminous efficiency–current density–luminance (LE–J–L) characteristics. | ||
Conclusions
Green-emitting hyperbranched polymers with a bipolar 3,6-carbazole-co-2,8-dioctyl-3,7- dibenzothiophene-S,S-dioxide [Cz-co-DOSO] as the branch and fac-tris(2-phenylpyridine) iridium [Ir(ppy)3] as the core were synthesized by Suzuki polycondensation. The Cz-co-DOSO branch with a triplet energy level above 2.5 eV can effectively prevent energy feedback from the phosphorescent core. A maximum luminous efficiency of 50.5 cd A−1 and a maximum external quantum efficiency of 15.3% were obtained for the single layer device based on PCzDOSO40Ir3, which were improved by over 13 times compared with PCzIr3. The hole- and electron-only devices show that the hole and electron flux are well balanced, which demonstrates that PCzDOSO40Ir3 is a bipolar polymer with a balanced charge carrier transport.Experimental
Materials
Commercially available reagents were used without further purification, unless stated otherwise. Toluene and THF were purified by routine procedures and distilled in dry argon atmosphere before used. N-(2-Ethylhexyl)-3,6-dibromocarbazole (1), N-(2-ethylhexyl)-3,6-bis-(4,4,5,5-tetra-methyl-1,3,2-dioxaborolan-2-yl)carbazole (2),16fac-tri(2-(3-bromo-phenyl)-pyridine)iridium (3)11 and 2,8-dioctyldibenzothiophene-S,S-dioxide13a were prepared according to our previous reports.Measurements
NMR spectra were recorded on a Bruker DRX 300 spectrometer at 300 MHz for 1H and at 75 MHz for 13C in deuterated chloroform with tetramethylsilane as a reference. The molecular weight of the polymers was determined by waters GPC 2410 using THF as the eluent. Cyclic voltammetry (CV) data were measured on a CHI800C electrochemical workstation using Bu4NPF6 (0.1 M) in acetonitrile as the electrolyte at a scan rate of 50 mV s−1 at r.t. under the protection of argon. Elemental analyses were performed on a Vario EL elemental analysis instrument (Elementar Co.). Thermogravimetric analyses (TGA) were performed on a Netzsch TG 209 at a heating rate of 20 °C min−1. The differential scan calorimetry (DSC) were measured on a Netzsch DSC 204 under N2 flow at a heating rate of 10 °C min−1.The iridium content was determined by using a Philips (Magix PRO) sequential X-ray fluorescence spectrometer (XRF), with a rhodium tube operated at 60 kV and 50 mA, a LiF 200 crystal and a scintillation counter. UV-Vis absorption spectra were recorded on a HP 8453 spectrophotometer. The PL spectra were recorded on a Hitachi F-4500 fluorescence spectrophotometer. The current denstiy (J)–voltage (V)–luminance (L) characteristics were collected by using a Keithley 236 source measurement unit and calibrated silicon photodiode. The EQE of EL was collected by measuring the total light output in all directions in an integrating sphere with calculated photodiodes (IS-080, Labsphere). The luminance was calibrated by a PR-705 Specra Scan Spectrophotometer (Photo Research), with simultaneous acquisition of the EL spectra and CIE coordinates, driven by Keithley model 2400 voltage–current source.Device fabrication
The fabrication process of the light-emitting diodes followed a standard procedure described below. The ITO-coated glass substrate underwent a wet-cleaning course in an ultrasonic bath sequentially in acetone, isopropanol, detergent, deionized water and isopropanol. After a 40 nm thick buffer layer of PEDOT:PSS (Baytron P4083) was spin-cast on the pre-cleaned ITO substrate and baked in a vacuum oven at 80 °C for 12 h, a 80 nm thick polymer layer was spin-cast from xylene solution on the top of the PEDOT:PSS and annealed at 100 °C for 20 min. Profilometry (Tencor Alfa-Step 500) was used to determine the thickness of the films. Typically, a thin layer of CsF (1.5 nm thick) with a 100 nm thick Al capping layer were deposited through a shadow mask (defined active area of 0.19 cm2) in a chamber with a basic pressure of < 3 × 10−4 Pa. The hole- and electron-only devices were used as ITO/PEDOT:PSS/polymer(165 nm)/MoO3(10 nm)/Al and ITO/Al(30 nm) /polymer(165 nm)/Ca(9 nm)/Al, respectively. The layer thickness was monitored upon deposition by a crystal thickness monitor (Sycon). Except the fabrication of PEDOT:PSS layer, all the processes were carried out in a N2 atmosphere dry-box (Vacuum Atmosphere Co.)Preparation of 3,7-dibromo-2,8-dioctyldibenzothiophene-S,S-dioxide (4)
2,8-Dioctyldibenzothiophene-S,S-dioxide (17.6 g, 40 mmol), trifluoroacetic acid (150 mL) were added into a single-necked flask (250 mL), and then bromine (6.2 mL, 120 mmol) was added dropwise with stirring. The mixture was stirred at room temperature in dark for 24 h. Then the mixture was poured into ice-water, extracted with CH2Cl2, washed with brine, and dried over MgSO4. After removal of the organic solvent, the crude product was purified by silica chromatography to give the white product with a yield of 72%.1H NMR (300 MHz, CDCl3) δ (ppm): 7.93 (s, 2 H), 7.59 (s, 2 H), 2.84 (t, J = 5.7 Hz and 6.0 Hz, 4H), 1.67 (m, 4H), 1.35-1.28 (br, 20H), 0.91 (t, J = 5.1 Hz, 6H). 13C NMR (75 MHz, DMSO) δ (ppm): 148.94, 136.57, 130.20, 126.50, 126.12, 122.65, 36.82, 31.82, 29.65, 29.39, 29.32, 29.18, 22.62, 14.06. Elemental Anal. Calcd for C28H38O2SBr2: C, 56.19%, H, 6.35%, O, 5.35%, S, 5.36%. Found: C, 56.21%, H, 6.42%, O, 5.43%, S, 5.32%.General procedures of Suzuki polycondensation, taking PCzDOSO40Ir3 as an example
N-(2-Ethylhexyl)-3,6-dibromocarbazole (1) (0.024 g, 0.055 mmol), N-(2-ethylhexyl)-3,6-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)carbazole (2) (0.2655 g, 0.5 mmol), fac-tri(2-(3-bromo-pheny)-pyridine) iridium (3) (0.0267 g, 0.03 mmol), 3,7-dibromo-2,8-dioctyldibenzothiophene-S,S-dioxide (4) (0.2392 g, 0.4 mmol), Pd(OAc)2 (3 mg) and tricyclohexylphosphine (6 mg) were dissolved in toluene (15 mL), stirred and heated to 90 °C, then Et4NOH(35wt%) aqueous solution (2 mL) was added. The mixture was stirred for 40 h under argon atmosphere. Then the polymers were capped by adding 0.02 g of N-(2-ethylhexyl)-3,6-bis (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)carbazole followed by stirring for 12 h, and by adding 0.5 mL of bromobenzene followed by heating for another 12 h. The whole mixture was poured into methanol. The precipitated polymers were recovered by filtration and purified by silica column chromatography with toluene to remove the small molecules and catalyst residue. A yellowish powder was isolated (yield 60%). 1H NMR (300 MHz, CDCl3), δ (ppm): 8.06 (br s, Ar H), 7.82 (br s, Ar H), 7.78(br m, Ar H), 7.76 (br m, Ar H),7.51–7.44(br m, Ar H), 4.26 (br s, N H), 2.80 (br s, CH2), 1.42–1.12 (br m, CH2), 1.00–0.75 (br m, CH3). Anal. Found (%): C, 80.03; H, 8.746; N, 2.631; S, 3.903.Acknowledgements
This research was financially supported by the State Key Basical Research Project of China (Nos. 2009CB623600 and 2009CB930604) and the National Nature Science Foundation of China (Nos. U0634003 and 21074038). We thank Prof. Chuluo Yang and Mr. Shaolong Gong at Wuhan University for the measurement of phosphorescent spectra.References
- (a) M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS; (b) E. Holder, B. M. W. Langeveld and U. S. Schubert, Adv. Mater., 2005, 17, 1109 CrossRef CAS; (c) V. Marin, E. Holder, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 618 RSC; (d) C. Ulbricht, B. Beyer, C. Friebe, A. Winter and U. S. Schubert, Adv. Mater., 2009, 21, 4418 CrossRef CAS.
- (a) P. T. Chou and Y. Chi, Chem.–Eur. J., 2007, 13, 380 CrossRef CAS; (b) Y. Kawamura, K. Goushi, J. Brooks, J. J. Brown, H. Sasabe and C. Adachi, Appl. Phys. Lett., 2005, 86, 071104 CrossRef.
- (a) X. Gong, M. R. Robinson, J. C. Ostrowski, D. Moses, G. C. Bazan and A. J. Heeger, Adv. Mater., 2002, 14, 581 CrossRef CAS; (b) C. Jiang, W. Yang, J. Peng, S. Xiao and Y. Cao, Adv. Mater., 2004, 16, 537 CrossRef CAS; (c) H. B. Wu, J. H. Zou, F. Liu, L. Wang, A. Mikhailovsky, G. C. Bazan, W. Yang and Y. Cao, Adv. Mater., 2008, 20, 696 CrossRef CAS; (d) Y. Byun, Y.-Y. Lyu, R. R. Das, O. Kwon, T.-W. Lee and Y. J. Park, Appl. Phys. Lett., 2007, 91, 211106 CrossRef.
- (a) J. Pina, J. S. de Melo, H. D. Burrows, A. P. Monkman and S. Navaratnam, Chem. Phys. Lett., 2004, 400, 441 CrossRef CAS; (b) Z. Chen, C. Y. Jiang, Q. L. Niu, J. B. Peng and Y. Cao, Org. Electron., 2008, 9, 1002 CrossRef CAS.
- (a) X. Chen, J. Liao, Y. Liang, M. O. Ahmed, H. E. Tseng and S.-A. Chen, J. Am. Chem. Soc., 2003, 125, 636 CrossRef CAS; (b) J. X. Jiang, C. Y. Jiang, W. Yang, H. Y. Zhen, F. Huang and Y. Cao, Macromolecules, 2005, 38, 4072 CrossRef CAS; (c) N. R. Evans, L. S. Devi, C. S. K. Mak, S. E. Watkins, S. I. Pascu, A. Kohler, R. H. Friend, C. K. Williams and A. B. Holmes, J. Am. Chem. Soc., 2006, 128, 6647 CrossRef CAS; (d) A. J. Sandee, C. K. Williams, N. R. Evans, J. E. Davies, C. E. Boothby, A. Köhler, R. H. Friend and A. B. Holmes, J. Am. Chem. Soc., 2004, 126, 7041 CrossRef CAS; (e) H. Y. Zhen, C. Y. Jiang, W. Yang, J. X. Jiang, F. Huang and Y. Cao, Chem.–Eur. J., 2005, 11, 5007 CrossRef CAS; (f) H. Y. Zhen, C. Luo, W. Yang, W. Y. Song, B. Du, J. X. Jiang, C. Y. Jiang, Y. Zhang and Y. Cao, Macromolecules, 2006, 39, 1693 CrossRef CAS.
- (a) M. Sudhakar, P. I. Djurovich, T. E. Hogen-Esch and M. E. Thompson, J. Am. Chem. Soc., 2003, 124, 7796 CrossRef; (b) R. J. Holmes, S. R. Forrest, Y. J. Tung, R. C. Kwong, J. J. Brown, S. Garon and M. E. Thompson, Appl. Phys. Lett., 2003, 82, 2422 CrossRef CAS; (c) S.-P. Huang, T.-H. Jen, Y.-C. Chen, A.-E. Hsiao, S.-H. Yin, H.-Y. Chen and S.-A. Chen, J. Am. Chem. Soc., 2008, 130, 4699 CrossRef CAS; (d) I. Avilov, P. Marsal, J. L. Bredas and D. Beljonne, Adv. Mater., 2004, 16, 1624 CrossRef CAS; (e) T. Tsuzuki and S. Tokito, Adv. Mater., 2007, 19, 276 CrossRef CAS.
- (a) A. V. Dijken, J. J. A. M. Bastiaansen, N. M. M. Kiggen, B. M. W. Langeveld, C. Rothe, A. Monkman, I. Bach, P. Stössel and K. Brunner, J. Am. Chem. Soc., 2004, 126, 7718 CrossRef; (b) K. Brunner, A. V. Dijken, H. Börner, J. J. A. M. Bastiaansen, N. M. M. Kiggen and B. M. W. Langeveld, J. Am. Chem. Soc., 2004, 126, 6035 CrossRef CAS; (c) Y. C. Chen, G. S. Huang, C. C. Hsiao and S.-A. Chen, J. Am. Chem. Soc., 2006, 128, 8549 CrossRef CAS.
- Y. M. You, S. H. Kim, H. K. Jung and S. Y. Park, Macromolecules, 2006, 39, 349 CrossRef CAS.
- H. Y. Zhen, J. Luo, W. Yang, Q. L. Chen, L. Ying, J. H. Zou, H. B. Wu and Y. Cao, J. Mater. Chem., 2007, 17, 2824 RSC.
- (a) P. L. Burn, S.-C. Lo and I. D. W. Samuel, Adv. Mater., 2007, 19, 1675 CrossRef CAS; (b) T. Guo, R. Guan, J. H. Zou, J. Liu, L. Ying, W. Yang, H. B. Wu and Y. Cao, Polym. Chem., 2011, 2, 2193 RSC.
- R. Guan, Y. H. Xu, L. Ying, W. Yang, H. B. Wu, Q. L. Chen and Y. Cao, J. Mater. Chem., 2009, 19, 531 RSC.
- (a) J. Liu, J. H. Zou, W. Yang, H. B. Wu, C. Li, B. Zhang, J. B. Peng and Y. Cao, Chem. Mater., 2008, 20, 4499 CrossRef CAS; (b) Y. Y. Li, H. B. Wu, J. H. Zou, L. Ying, W. Yang and Y. Cao, Org. Electron., 2009, 10, 901 CrossRef CAS; (c) S. M. King, I. I. Perepichka, I. F. Perepichka, F. B. Dias, M. R. Bryce and A. P. Monkman, Adv. Funct. Mater., 2009, 19, 586 CrossRef CAS; (d) I. I. Perepichka, I. F. Perepichka, M. R. Bryce and L.-O. Pålsson, Chem. Commun., 2005, 3397 RSC.
- (a) J. Liu, S. J. Hu, W. Zhao, Q. H. Zou, W. Luo, W. Yang, J. B. Peng and Y. Cao, Macromol. Rapid Commun., 2010, 31, 496 CrossRef CAS; (b) K. T. Kamtekar, H. L. Vaughan, B. P. Lyons, A. P. Monkman, S. U. Pandya and M. R. Bryce, Macromolecules, 2010, 43, 4481 CrossRef CAS.
- S. Janietz, D. D. C. Bradley, M. Grell, C. Giebeler, M. Inbasekaran and E. P. Woo, Appl. Phys. Lett., 1998, 73, 2453 CrossRef CAS.
- Y. T. Tao, Q. Wang, C. L. Yang, K. Zhang, Q. Wang, T. T. Zou, J. G. Qin and D. G. Ma, J. Mater. Chem., 2008, 18, 4091 RSC.
- J. Huang, Y. H. Niu, W. Yang, Y. Q. Mo, M. M. Yuan and Y. Cao, Macromolecules, 2002, 35, 6080 CrossRef CAS.
- (a) C.-L. Lee, N.-G. Kang, Y.-S. Cho, J.-S. Lee and J.-J. Kim, Opt. Mater., 2002, 21, 119 CrossRef; (b) S. Tokito, M. Suzuki, F. Sato, M. Kamachi and K. Shirane, Org. Electron., 2003, 4, 105 CrossRef CAS; (c) M. Suzuki, S. Tokito, F. Sato, T. Igarashi, K. Kondo, T. Koyama and T. Yamaguchi, Appl. Phys. Lett., 2005, 86, 103507 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |