Medium/high temperature water gas shift reaction in a Pd–Ag membrane reactor: an experimental investigation
Adele
Brunetti
a,
Enrico
Drioli
ab and
Giuseppe
Barbieri
*a
aNational Research Council–Institute on Membrane Technology (ITM–CNR) Via Pietro BUCCI, c/o The University of Calabria, cubo 17C, 87036, Rende CS, Italy. E-mail: g.barbieri@itm.cnr.it; Fax: +39 0984 402103; Tel: +39 0984 492029
bThe University of Calabria - Department of Chemical Engineering and Materials, cubo 44A Via Pietro BUCCI, 87036, Rende CS, Italy
First published on 3rd November 2011
Abstract
In the hydrogen production cycle the syngas stream coming out from reformers or coal gasification plants contains a large percentage of H2 (ca. 50%) and CO (ca 45%). In fact, it is systematically upgraded by a water gas shift reaction (WGS) in order to convert the CO present and, at the same time, to produce further hydrogen. Water gas shift is an exothermic thermodynamics limited reaction and the presence of products like H2 further depletes the CO conversion achievable. In traditional applications this upgrading stage is usually performed in two reaction stages: firstly, the syngas stream is fed to a reactor operated in a high temperature range (300–400 °C) to exploit the advantages offered by a fast kinetics; then, the outlet stream of the first reactor is fed, after cooling, in a low temperature stage, for the benefit of the thermodynamics. This final stream is, thus, fed to a separation unit to recover the hydrogen from the rest of the gaseous stream. In this work the upgrading of a syngas stream is experimentally investigated in a Pd-based membrane reactor (MR) operated in the medium/high temperature range (340–375 °C). The advantages on the MR performance offered by fast kinetics and permeation rate have been analyzed as a function of the feed pressure (up to 1100 kPa), feed molar ratio, and gas hourly space velocity (GHSV). The values of these variables used in the experiments are closer to those used in the industrial applications. The MR performance, compared with the ones of a traditional reactor (TR) operated in the same conditions, was evaluated also in terms of volume index which evidences the lower catalyst volume required by an MR for achieving the same conversion of a TR. Owing to the fast kinetics and permeation rate, the limit due to the presence of product (i.e., H2) in the feed stream added to that imposed by the thermodynamics were successfully overcome with the MR operated in the high temperature range. A CO conversion significantly higher than the thermodynamics upper limit of a TR was achieved, also at high values of GHSV and less than 30% of the reaction volume of a TR was required for achieving a conversion equal to 90% of the traditional reactor equilibrium conversion.
Introduction
In the last decade, the energy demand has been growing by 1.2% per year and fossil fuels still maintain a production share of ca. 75%. However, the ever more severe problems connected to a sustainable growth and to a lower environmental impact lead to the necessity to free energy production from oil and natural gas as primary energy sources. Consequently there is a need for the development of clean technologies, designed to enhance both the efficiency and environmental acceptability of energy production, storage and use, in particular for power generation.1 Among these technologies, the exploitation of light hydrocarbons for hydrogen production is surely the main realistic energy source, since it allows both power generation and environment-friendly fuel production.At present, 96% of hydrogen is directly produced from fossil fuels and about 4% is produced indirectly by using electricity generated through them.2 The stream coming out from a reformer or a coal gasification plant contains around 50% hydrogen (on a dry basis) that must be recovered and between 40–45% CO that, usually, is reduced in an upgrading stage, producing more hydrogen at the same time. The upgrading of reformate streams is the water gas shift (WGS), exothermic reaction characterized by no variation of the mole number.
| CO + H2O = CO2 + H2⋯ΔH0298 = −41 kJ mol−1 |
Therefore, CO conversion is thermodynamically favoured by low temperatures and pressure does not affect the conversion in traditional reactors. On the contrary, temperature promotes the WGS kinetics and pressure also favours the selective permeation of hydrogen.
In traditional applications, this conversion process is based on multi-stage CO–shift reactors: the first, based on Fe2O3–Cr2O33,4,5 catalyst is operated at high temperatures (about 350–400 °C) to take advantage of the high reaction rate, and converts a large part of the carbon monoxide to give hydrogen and CO2; the other, operated at a low temperature (around 220–300 °C) by using CuO–ZnO,3,5,6 refines the carbon monoxide conversion, thus lowering the CO final concentration (less than 1% molar).7 The CuO–ZnO catalysts undergo sintering at a higher temperature and present a lower reaction rate with respect to Fe2O3–Cr2O3 catalysts; therefore, usually the typical GHSV (gas hourly space velocity) of the second stage is ca. tenfold lower than the high temperature reactor. This H2 rich stream coming out from the last reactor is then fed to a separation stage to separate H2 from the other gases. It should be pointed out that the new utilization of H2 as feed in fuel cells for mobile and stationary power sources requires the anode inlet gas to have a CO concentration lower than 10–20 ppm8 to avoid catalyst poisoning with consequent drops in the fuel cell efficiency. Hence, the purification step of the H2 produced from hydrocarbon must be very efficient to fulfil these requirements. For this reason in some cases, another reaction unit, CO selective oxidation, is added to convert CO in CO2.
One of the main challenges in the next few years will be not only the identification of new technologies able to provide a better exploitation of raw materials, but also the reduction of the reaction/separation/purification stages. This means lower footprint area occupied by the whole plant, less auxiliary devices required, reduction of the energetic load, all fundamental aspects to be taken into account in the new design of hydrogen production processes. A promising approach for concretizing these technological aspects in the field of hydrogen production is the use of MRs, combining the reaction and H2 separation by means of a selective membrane. Many studies are now focused on the analysis of MR performances where WGS reaction is carried out. In this case, the presence of the membrane allows the recovery of a hydrogen reach/pure stream which does not require any further separation. Moreover, the removal of the H2, reaction product from the reaction volume shifts the reaction toward a further conversion. This means the possibility of having an intensified process with a reduced plant size and a higher yield. The traditional process can be thus redesigned as more compact and efficient, pursuing the logic of the Process Intensification strategy,1,2 which is an innovative methodology for process and plant design. This leads to a new design philosophy to achieve significant reductions (by factors of 10 to 100 or more) in plant volume at the same production capacity and to improve overall efficiency. One MR operating at high temperature can replace9,10 the two reactors of the conventional process while giving the same final conversion. An analysis by means of a mathematical model and simulations demonstrated a dramatic reduction of reaction volume when an Fe–Cr-based catalyst is used. Among the various membrane types, Pd-alloy membranes are the most used in applications such as dehydrogenation reactions,11,12,13 because of their infinite H2 selectivity.14 The hydrogen removal gives several advantages with respect to the traditional operations:
• Depleting the reverse reaction rate due to the lower H2 concentration
• Increasing the residence time of reactants
• Exceeding the thermodynamic equilibrium of a traditional reactor
• Pressure was demonstrated15 to have a positive effect on conversion in an MR for reactions characterised by an increase of the mole number such as, e.g., methane steam reforming as well as WGS (with no variation in the number of moles) even though, in a TR, it depletes or does not affect the conversion of methane steam reforming and WGS, respectively.
In the past, palladium-alloy membranes were already successfully used for hydrogen production/separation also for the WGS step.11,14,16–23 In some cases, the interesting results achieved at laboratory level concretized in various patents.24–29 Among them, United Technologies Corp.24 patented the use of a WGS MR, comprising a WGS reaction region and a permeate volume, separated by an H2–separation membrane which allows H2 formed over a catalyst in the reaction region to be passed selectively to the permeate region for delivery to a point of use (such as the fuel cell of a fuel cell power plant). Also the General Electric Company26 patented a polygeneration system including (a) a syngas generator for producing a syngas, (b) a syngas enrichment unit for removing undesired species from the syngas for producing an enriched syngas and (c) a syngas utilization system that utilizes the enriched syngas to produce useful products. In some embodiments, the polygeneration system includes MR, catalytic burner and power generation unit. However, in the majority of these studies the WGS was carried out in the low or medium temperature range (180–250 and 250–320 °C, respectively) by using CuO-based catalysts.
Only recently, Augustine et al.30 have performed the WGS in a catalytic MR operating at elevated temperatures (ca.450 °C) and pressures (ca 14 bar). However, they carried out the experiments with a composite Pd-membrane prepared in lab and exhibiting a finite, even if high (ca.450), selectivity toward hydrogen. Moreover, the GHSVs investigated ranged up to 6,021 h−1.
This work presents an analysis of the performance of a Pd–Ag MR for the WGS reaction carried out in the high temperature range (340–375 °C), by using a commercial Pd–Ag self-supported membrane, which exhibited infinite hydrogen selectivity. In the traditional operation, the reaction operated in this temperature range is strongly depleted by thermodynamics constrains. The MR overcomes the limitation imposed by the thermodynamics to TR due to hydrogen removal promoted by pressure and, hence, the fast kinetics characterizing the Fe–Cr based catalysts can be successful exploited. CO conversion, hydrogen production as well as reaction volume and catalyst amount required were analysed as a function of feed pressure, temperature, H2O/CO feed molar ratio and GHSV. The latter was varied from 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 021 to 13300 h−1 in order to be closer to the real industrial operating conditions.
021 to 13300 h−1 in order to be closer to the real industrial operating conditions.
Materials and methods
The experiments were carried out in a tube in-tube MR (Fig. 1) where the outer tube is a stainless steel shell, whereas the inner tube is the Pd-alloy self-supported membrane, blind on one side. Data about the membrane and MR are summarized in Table 1. | ||
| Fig. 1 Scheme of the Pd-based Membrane reactor. | ||
| Membrane | |
|---|---|
| Membrane type | Pd–Ag - Commercial (Goodfellow) Self-supported |
| Thickness | 100 micrometres |
| Area available for hydrogen permeation | 2 cm2 |
| Outer Diameter | 1 mm |
| Length | 65 mm |
| Membrane Reactor (the catalyst was packed in the annular volume) | |
| Inner Diameter | 6 mm |
| Length | 70 mm |
| Catalyst type | CeO2/CrO based catalyst KATALCO® (Johnson Matthey) |
| Catalyst weight | 11.3 g |
A CeO2/CrO-based catalyst named KATALCO® (supplied by Johnson Matthey) was packed in the shell side, where the reaction occurs and the permeated stream is recovered in the core of the tubular membrane.
The experimental apparatus used in the reaction tests is schematically shown in Fig. 2. The MR was placed in a temperature (PID) controlled electric furnace. A mass flow controller (Brooks Instrument 5850S) was used for feeding the gaseous mixture, and an HPLC pump (Dionex P680A) was used to feed the water. A heating coil for vaporizing the water was put into the furnace, close to the MR. The flow rates of the outlet streams were measured by means of bubble soap flow-meters. The temperature was measured by using a thermocouple positioned on the middle of the reactor shell. The retentate and permeate streams compositions were analysed by means of a gas chromatograph (Agilent 6890N) with two parallel analytical lines. Each line is equipped with two columns: an HP-Plot-5A (for separating permanent gases such as H2, N2 and CO) and an HP-Poraplot-Q (for other species) and a TCD. This allowed contemporary analysis of the retentate and permeating streams.31
 | ||
| Fig. 2 Scheme of the experimental laboratory-scale plant. MFC: mass flow controller; GC: gas chromatograph. | ||
Generally, as in this work, the permeating flux of H2 through Pd-alloy dense membranes can be described by Sievert’s law (Eqn (1)) since these membranes are utilised in a temperature range where the diffusion in the metal bulk is the rate determining step. In this case, the hydrogen permeating flux is a linear function of the driving force, which is given by the difference of the square root of the H2 partial pressure on both membrane sides.
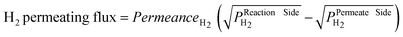 | (1) |
CO conversion of TR and MR was calculated using the Eqn (2), including CO and CO2 present in the feed and retentate streams.18 In particular it was calculated (using the molar flow rate, F) as the mean value of CO2 produced (lower value) and CO present in the retentate (upper value), the difference between them being the carbon balance.
 | (2) |
The MR capability to recover hydrogen was quantified using the Eqn (3)18 as the H2 fraction permeated through the membrane with respect to the whole H2 present in the reaction side, given by the sum of hydrogen produced by reaction and that present in the stream feeding the MR itself.
 | (3) |
The results obtained with the experiments have also been expressed in terms of Volume Index, Eqn (4).32 It is important to evaluate the MR performance in terms of new metrics for process intensification since gives an immediate idea of the gain offered by new reactor model with respect to TR. It is the ratio of the reaction volumes required for achieving the same conversion. It ranges 0 to 1 and the gain is much relevant as much it is close to 0.
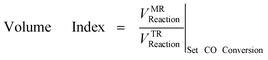 | (4) |
The operating conditions used for the experiments are reported in Table 2. A syngas stream, simulating the composition of a stream coming out by a reformer, was upgraded in the temperature range 340–375 °C analyzing, in particular, the effects of the GHSV (Equ. 5) and the feed pressure. GHSV is a variable indicating the inverse of the residence time of the reactant in the reactor or on the catalyst phase. A low GHSV corresponds to a high residence time and, thus, favours the conversion. On the contrary, a high GHSV implies a reduced residence time and, thus, a lower conversion of reactants. However, industrially a high GHSV value is desired since it means the possibility of treating a huge amount of reactants with a low catalyst amount in a small reactor.
 | (5) |
| Temperature | 340–375 °C | |
| Feed Pressure | 7.5–11 bar | |
| Permeate Pressure | 1 bar | |
| H2O/CO feed molar ratio | 0.7; 1; 2 | |
| GHSV (gas hourly space velocity) | 6021; 10035; 13300 h−1 |
|
| Feed composition (dry) (%) | CO:H2:CO2:N2 = 45:50:4:1 |
|
| No sweep gas was used | ||
In this work, it was chosen to explore a wide range of GHSV to analyze the performance of the MR under operating conditions closer to the real industrial application. The role of driving force, responsible for the permeation, was assigned to the feed pressure. For this reason, no sweep gas was used during the experiments and the permeate side was kept constant at atmospheric pressure to obtain a pure hydrogen stream on the latter side.
The integrity of the membrane and its infinite hydrogen selectivity was verified by feeding nitrogen at 5 bar and confirming the absence of any nitrogen flux on the permeate side.
Results and discussion
Hydrogen transport through the membrane
The H2 permeating flux through the Pd–Ag membrane was measured by feeding pure gas and the permeation membrane properties (flux and permeance) as a function of the permeation driving force were evaluated before carrying out the reaction experiments.As expected, a linear dependence of the H2 flux as a function of the driving force was observed at both the temperatures investigated (Fig. 3), therefore it was confirmed that hydrogen flux follows Sievert's law and a constant permeance value can be assumed for each temperature (Table 3).
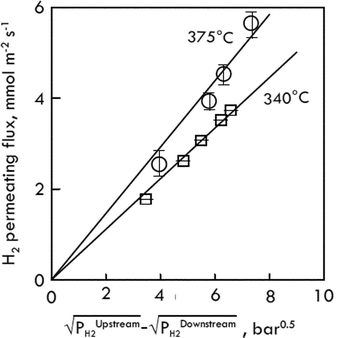 | ||
| Fig. 3 Hydrogen permeating flux as a function of driving force measured (symbols) at 340 and 375 °C and Upstream pressure up to 11 bar. Lines: linear regression through the axes origin. Pd–Ag membrane. | ||
| T/°C | Permeance mmol m−2 s−1 bar−0.5 | Permeability nanomol m m−2 s−1 bar−0.5 |
|---|---|---|
| 340 | 1.76±0.004 | 180±0.4 |
| 375 | 2.31±0.056 | 230±6 |
Barbieri et al. (2008)18 showed that a there is no inhibition to hydrogen permeation owing to the carbon monoxide adsorption on the metal membrane surface at a temperature higher than 300–350 °C. The species concentration gradients close to the membrane surfaces for such a thick membrane (100 micron) was demonstrated (Caravella et al., 200933) to be very low. Therefore, there is no deviation from Sievert's law neither due to the CO inhibition nor concentration gradients in the films close to the membrane (named concentration polarization).
Reaction investigations
As previously mentioned,9,10 the aim of this work was the demonstration of the suitability of an MR for performing the HT-WGS reaction, since in general, much better performances than TR ones are obtained. Indeed, the reaction measurements were carried out at medium/high temperature and a wide range of GHSV was explored to analyze the performance of the MR under operating conditions closer to the industrial application. The performance of a Pd–Ag MR is significantly favoured by the pressure difference (permeation driving force) between the two membrane sides. The feed pressure acts positively on the CO conversion and on H2 recovery, allowing more hydrogen to be recovered in the permeate, since permeate pressure is atmospheric. Even though the feed pressure does not influence the reaction from a thermodynamic point of view, the removal of hydrogen from the reaction sides shifts the reaction toward further formation of products. Fig. 4 shows CO conversion as a function of the feed pressure, measured at 340 and 375 °C for different GHSV values. The CO conversion increased with the feed pressure for both the temperatures and all the GHSVs investigated and, it was in all the cases, significantly higher than TREC that, as expected, remained constant over the whole pressure range. As the CO conversion increased, the H2 recovery increased too (Fig. 5): it being the H2 fraction collected in the permeate with respect to that totally fed/produced into MR (Eqn 3). In all the cases more than 40% of the hydrogen present in the reactor was continuously collected as pure in the permeate: at 375 °C, it reached a value slightly higher than 60% at 11 bar and GHSV = 6021 h−1. Both the CO conversion and the H2 recovery range were determined by the GHSV. The latter indicates the reverse of the contact time between the reactants and the catalyst (Eqn 5), therefore the higher the GHSV the lower the CO conversion (Fig. 6).
 | ||
| Fig. 4 CO conversion as a function of feed pressure for different values of GHSV, measured at 340 and 375 °C. | ||
 | ||
| Fig. 5 H2 recovery as a function of feed pressure for different values of GHSV, measured at 340 and 375 °C. | ||
 | ||
| Fig. 6 CO conversion as a function of GHSV at different values of feed pressure, measured at 340 and 375 °C. | ||
The CO conversion decreased from 75 to 59% as the GHSV increased from 6000 to 13300 h−1 at 340 °C and 8.8 bar. It happened also at 11 bar, where the CO conversion dropped down from 83 to 68%. The same trends were observed at 375 °C. In any case, also at the highest GHSV, the CO conversion obtained by using the Pd–Ag MR was 2–3 times higher than that measured with the TR in the same operating conditions and always exceeded the TREC, the highest value achievable in a TR. As a consequence of the lower CO conversion, the H2 percentage removed from the reaction side and recovered in the permeate side decreased as the GHSV increased (Fig. 7). A lower CO conversion meant, in other words, less hydrogen production or, rather, a lower H2 partial pressure on the reaction side. This is traduced in a lower permeation driving force and, thus, in less H2 recovered in the permeate.
 | ||
| Fig. 7 H2 recovery as a function of GHSV at different values of feed pressure, measured at 340 and 375 °C. | ||
Fig. 8 highlights the effect of the temperature on the CO conversion for different values of feed pressure and GHSV, when both MR and TR are compared. From a general point of view the high temperature has a competitive effect on the reactor performance. Thermodynamically, the high temperature depletes the WGS since an exothermic reaction; nevertheless, both the reaction kinetics and the H2 permeation are favoured, since they follow an Arrhenius law. Therefore, as can be seen in Fig. 8, as the temperature increased in the MR the CO conversion increased too; the advantages induced on kinetics and H2 permeation were more significant than the depletion due to the thermodynamics. The contrary happened in the TR, where no permeation occurred, indeed, the thermodynamic constraints became dominant over the kinetics assets. For both the temperature values investigated, the conversion achieved in the MR was always ca. 3 to 5 times higher that than of the TR, always exceeding the TREC. As a combination of the positive effect supplied by the feed pressure and the high temperature as well, the highest CO conversion (87%) was obtained at 11 bar and 375 °C. Since the H2 recovery depends on the permeating capacity of the system; the higher the temperature and feed pressure the higher its value (Fig. 7). At 11 bar, 6021 h−1 and 375 °C, around 63% of the H2 fed or produced by reaction was recovered as pure in the permeate of the MR.
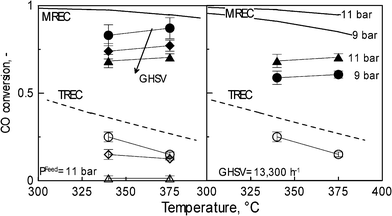 | ||
| Fig. 8 CO conversion as a function of temperature for different values for GHSV equal to 6021, 10035, 13300 h−1 (left side) and feed pressure (right side). | ||
As an indicator of the system reduced size, the Volume Index was calculated to evaluate the gain offered by the use of an MR in terms of reaction volume with respect to a simple TR. The volume index is a decreasing function of the feed pressure (Fig. 9). At 6021 h−1 and 340 °C, the reaction volume required by the MR to achieve a final conversion of ca. 33% (corresponding to 90% of the TREC), was around 29% of that of a TR and this value dropped down to 16.3% when increasing the feed pressure at 11 bar. Furthermore, at the highest temperature of 375 °C, the volume index was still reduced, passing from 0.22 to 0.16 at 9 bar and 11 bar, respectively. A high feed pressure and a high temperature, in fact, implying that more H2 permeates through the membrane and, thus, shifting the reaction towards further CO conversion, require a lesser amount of catalyst to achieve the desired set conversion. Nevertheless, the lowest reaction volume was reached at the lower GHSV, a significant gain was achieved by using MR also at the highest GHSV value. Table 4 summarizes the values of the catalyst volume required by an MR with respect to a TR as a function of the GHSV, calculated by the experimental data for the different temperatures and feed pressures. As it is highlighted, the catalyst amount or, in other words, MR reaction volume is in all cases lower than the TR one. In particular, at 13300 h−1and 375 °C, operating conditions much closer to the actual MT/HT-WGS industrial application, the catalyst required by MR operated with 9 bar of feed pressure was 35% of the TR one and it lowered down to 32% at a feed pressure equal to 11 bar.
 | ||
| Fig. 9 Volume Index as a function of feed pressure at 340 and 375 °C. | ||
Industrially, the H2O/CO feed molar ratio ranges up to 5 depending on the type of stream to be treated and on the target required5 it is significantly higher than the stoichiometric value of 1. A higher feed molar ratio increases the CO conversion strongly limited by the thermodynamics as well as the reaction rate. In addition, the syngas stream is often produced by steam reforming or partial oxidation of light hydrocarbons or coal gasification. In all these process an excess of water is used to improve the conversion and/or to avoid/reduce the coke formation, which deactivates the catalyst. Therefore, also an H2O/CO feed molar ratio of 2 was used even if very good results were obtained for the stoichiometric feed molar ratio in the MR (Fig. 10). A molar ratio of 0.7 was also considered, to analyze the performance of the MR under the worst condition.
 | ||
| Fig. 10 CO conversion as a function of molar ratio for 9 and 11 bar, measured at 340 °C and GHSV = 6021 h−1. | ||
The higher value of feed molar ratio gives a higher equilibrium conversion, therefore the TREC curve monotonically increases with feed molar ratio. As expected, an improvement in MR CO conversion was obtained reaching ca. 90% at 11 bar and a feed molar ratio equal to 2. Moreover, the measured CO conversion always exceeded the TREC for a very large amount for both the feed pressure values considered. However, the MR resulted much more convenient in relation to the feed molar ratio being close to the stoichiometric one. The MR CO conversion was more than 3 times higher than the TR one for a feed molar ratio of 1, whereas it became ca. 2.5 times higher operating with a molar ratio of 2.
Table 5 compares the performances of LT-WGS11 and MT/HT-WGS carried out in similar Pd–Ag MR. It must be pointed out that in both these temperature ranges the CO conversion achieved with MR was always higher than the TR one operated in the same conditions, as it is widely explained above in the text and in a previous work on medium temperature WGS in MR.11 Both these MRs reached high CO conversion and, in both of them, a fundamental role was played by the feed pressure, which is the driving force of the process. However, to achieve similar CO conversion values, in the MT-WGS, the GHSV used was significantly lower than the ones that could be used in the HT–WGS range. From a general point of view, this means that the WGS carried out in the HT range allows feed flow rates 2–4 times higher than the one operable in the MT stage to be treated using the same catalyst amount, or, in other words, to treat the same feed flow rate with a catalyst volume 2–4 times lower than the one required operating in the low temperature range. It might be highlighted that also in the LT stage there was already a gain in the catalyst volume required by the reaction, it being only 40% of the TR one and, thus, operation at the higher temperature offers an extra gain, further reducing the catalyst volume required.
| Low temperature WGS | |
|---|---|
| Feed Pressure = 6 bar; T = 300 °C | |
| GHSV, h−1 | CO conversion, - |
| 2500 |
0.89 |
| 3200 |
0.75 |
| Medium/high temperature WGS | |
|---|---|
| Feed Pressure = 11 bar; T = 375 °C | |
| GHSV, h−1 | CO conversion, - |
| 6000 |
0.87 |
| 10035 |
0.77 |
Conclusions
A syngas mixture was upgraded in one-stage using a Pd–Ag membrane by means of water gas shift (WGS) reaction carried out in the high temperature range. The fast kinetics together with the high permeation rate offered by the high temperature allowed the thermodynamics and the further limitations due to the H2 presence (50%) in the feed stream to be significantly exceeded. The measured CO conversion was 2–3 times higher than the one measured with the TR in the same operating conditions also for high values of GHSV: largely exceeding the equilibrium conversion of a TR. At the higher temperature of 375 °C and at 6021 h−1, the catalyst volume required by MR was only 15–20% of the TR to achieve the same conversion. Moreover, also at the higher GHSV values a significant gain was achieved by using MR. In particular, at 13300 h−1 and 375 °C, operating conditions much closer to actual medium/high temperature WGS industrial applications, the catalyst required by MR operated with 9 bar of feed pressure was 35% of the TR one and it lowered down to 31.7% at a feed pressure equal to 11 bar.
Finally, the present analysis shows a reduction of two reaction and one purification stage in only one unit, the MR, able to give CO conversion at least comparable with that of the whole shift process. This leads to a simpler process and implies important reductions in the footprint area occupied by the upgrading step. However, further efforts are required to identify the optimal operating conditions that enable to the MR to obtain the best performance.
List of symbols
| F | Molar flow rate, mol s−1 |
| J | Permeation flux, mol m2 s−1 |
| P | Pressure, Pa |
| T | Temperature, °C |
| V | Volume, m3 |
| VI | Volume Index (%) |
| GHSV | Gas hourly space velocity |
| MT/HT-WGS | Medium/high temperature water gas shift |
| LT-WGS | Medium temperature water gas shift |
| MR | Pd-based membrane reactor |
| MREC | Membrane reactor equilibrium conversion |
| PEMFC | Polymer electrolyte membrane fuel cell |
| PSA | Pressure swing adsorption |
| TR | Traditional reactor |
| TREC | Traditional reactor equilibrium conversion |
Acknowledgements
Dott.ssa Michela Musso (Johnson Matthey Catalysts - Market Manager Italy & France) is gratefully acknowledged for the catalyst supplied.References
- S. Wadhwani, A. K. Wadhwani, R. B. Agarwal, “Clean coal technologies – recent advances”, proceedings of the First International Conference on Clean Coal Technologies for Our Future, Chia Laguna (Sardinia), Italy, 200221-23 October 2002 Search PubMed.
- R. Kothari, D. Buddhi and R. L. Sawhney, Comparison of environmental and economic aspects of various hydrogen production methods, Renewable Sustainable Energy Rev., 2008, 12, 553 CrossRef CAS.
- F. Bustamante, R. M. Enick, A. V. Cugini, R. P. Killmeyer, B. H. Howard, K. S. Rothenberger, M. V. Ciocco, B. D. Morreale, S. Chattopadhyay and S. Shi, High-Temperature Kinetics of the homogeneous Reverse Water–Gas Shift Reaction, AIChE J., 2004, 50(5), 1028–1040 CrossRef CAS.
- R. L. Keiski, T. Salmi, P. Niemisto, J. Ainassaari and V. J. Pohjola, Stationary and transient kinetics of the high temperature water-gas shift reaction, Appl. Catal., A, 1996, 137, 349–370 CrossRef CAS.
- Ullman's Encyclopedia of Industrial Chemistry, 5th Completely Revised Edition. Edited by Barbara Elvers, Stephen Hawkins, and William Russey. VCH: New York, 1995. ISBN 3-527-20126-2.
- N. E. Amadeo, M. A. Laborde, Hydrogen production from the low temperature water gas shift reaction: kinetics and simulation of the industrial reactor.Int. J. Hydrogen Energy, 1995, 20, 949–958 Search PubMed.
- G. Raggio, A. Pettinau, A. Orsini, M. Fadda, D. Cocco, P. Deiana, M. L. Pelizza, M. Marenco, Second International Conference on Clean Coal Technologies for Our Future, Castiadas, Sardinia, Italy, 10–12 May 2005 Search PubMed.
- F. Barbir, PEM electrolysis for production of hydrogen from renewable energy sources, Sol. Energy, 2005, 78, 661 CrossRef CAS.
- G. Barbieri, A. -Brunetti, A. Caravella, E. Drioli, “Reattori a membrane per il processo di water gas shift ad un solo stadio”, Italian Patent, 2011, pending Search PubMed.
- G. Barbieri, A. Brunetti, A. Caravella and E. Drioli, Pd-based Membrane Reactors for one-stage Process of Water Gas Shift, RSC Adv., 2011, 1(4), 651–661 RSC.
- K. Babita, S. Sridhar and K. V. Raghavan, Membrane reactors for fuel cell quality hydrogen through WGSR – Review of their status, challenges and opportunities, Int. J. Hydrogen Energy, 2011, 36(11), 6671–6688 CrossRef CAS.
- J. Shu, B. P. A. Grandjean, A. Van Neste and S. Kaliaguine, Catalytic palladium-based membrane reactors: a review, Can. J. Chem. Eng., 1991, 69, 1036–1048 CrossRef CAS.
- A. Brunetti, G. Barbieri and E. Drioli, Upgrading of a syngas mixture for pure hydrogen production in a Pd-Ag membrane reactor, Chem. Eng. Sci., 2009, 64, 3448–3454 CrossRef CAS.
- F. A. Lewis, “Palladium Hydrogen System“, Academic Press, London, New York, 1967 Search PubMed.
- G. Marigliano, G. Barbieri and E. Drioli, Equilibrium conversion for a palladium membrane reactor. Dependence of the temperature and pressure, Chem. Eng. Process., 2003, 42, 231–236 CrossRef CAS.
- R. Dittmeyer, V. Hollein and K. Daub, Membrane reactors for hydrogenation and dehydrogenation processes based on supported palladium, J. Mol. Catal. A: Chem., 2001, 173, 135–84 CrossRef CAS.
- S. N. Paglieri and J. D. Way, Innovations in palladium membrane research, Sep. Purif. Rev., 2002, 31, 1–169 CrossRef CAS.
- G. Barbieri, A. Brunetti, G. Tricoli and E. Drioli, An innovative configuration of a Pd-based membrane reactor for production of pure hydrogen. Experimental analysis of Water Gas Shift, J. Power Sources, 2008, 182, 160–167 CrossRef CAS.
- A. Brunetti, G. Barbieri, E. Drioli, K.-H. Lee, B. Sea and D.-W. Lee, WGS Reaction in Membrane Reactor Using a Porous Stainless Steel Supported Silica Membrane, Chem. Eng. Process., 2007, 46, 119–126 CrossRef CAS.
- S. Battersby, M. C. Duke, S. Liu, V. Rudolph and J. C. Diniz da Costa, Metal doped silica membrane reactor: Operational effects of reaction and permeation for the water gas shift reaction, J. Membr. Sci., 2008, 316, 1-#x2013;2(15), 46–52 Search PubMed.
- D. Mendes, S. Sá, S. Tosti, J. M. Sousa, L. M. Madeira and A. Mendes, Experimental and modeling studies on the low-temperature water-gas shift reaction in a dense Pd–Ag packed-bed membrane reactor, Chem. Eng. Sci., 2011, 66(11), 2356–2367 CrossRef CAS.
- D. Mendes, V. Chibante, J.-M. Zheng, S. Tosti, F. Borgognoni, A. Mendes and L. M. Madeira, Enhancing the production of hydrogen via water–gas shift reaction using Pd-based membrane reactors, Int. J. Hydrogen Energy, 2010, 35(22), 12596–12608 CrossRef CAS.
- P. Pinacci, M. Broglia, C. Valli, G. Capannelli and A. Comite, Evaluation of the water gas shift reaction in a palladium membrane reactor, Catal. Today, 2010, 156, 3-#x2013;4(31), 165–172 Search PubMed.
- M. Gummalla, T. H. Vanderspurt, Y. She, Z. Dardas and B. Olsommer, “power plant with membrane water gas shift reactor system”, 2010, US20100104903 Search PubMed.
- H. W. Deckman, J. W. Fulton, J. M. Grenda and F. Hershkowitz, “Electric power generation with heat exchanged membrane reactor”, 2005, EP1294637 Search PubMed.
- W. Wei, “Polygeneration systems”, 2009, US20090084035 Search PubMed.
- D. Tsay, S. E. Weiss and T. C. Tsay, “Single stage membrane reactor for high purity hydrogen production”, 2007, US20070157517 Search PubMed.
- A. Lamm, T. Poschmann and J. Schaefer, “Apparatus for generating virtually pure hydrogen for fuel cells, 2005, US20050039401 Search PubMed.
- R. S. Willms and S. A. Birdsell, “Apparatus and method for simultaneous recovery of hydrogen from water and from hydrocarbons”, 2000, US6165438 Search PubMed.
- A. S. Augustine, Y. H. Ma and N. K. Kazantzis, High pressure palladium membrane reactor for the high temperature water–gas shift reaction, Int. J. Hydrogen Energy, 2011, 36(9), 5350–5360 CrossRef CAS.
- G. Barbieri, P. Bernardo, “Carbon Dioxide Capture and Storage in Deep Geologic Formations; Chapter 22 – GRACE: Experimental Evaluation of Hydrogen Production by Membrane Reaction”, pag. 385–407, 2005–Elsevier, Edited by David C. Thomas and Sally M.Benson Search PubMed(ISBN008044570).
- A. Brunetti, A. Caravella, G. Barbieri and E. Drioli, Simulation study of water gas shift in a membrane reactor, J. Membr. Sci., 2007, 306/1-#x2013;2, 329–340 Search PubMed.
- A. Caravella, G. Barbieri and E. Drioli, Concentration Polarization Analysis in Self-supported Pd-based Membranes, Sep. Purif. Technol., 2009, 66, 613–624, DOI:10.1016/j.seppur.2009.01.008.
| This journal is © The Royal Society of Chemistry 2012 |