Structured biotinylated poly(3,4-ethylenedioxypyrrole) electrodes for biochemical applications†
Thierry
Darmanin
a,
Hervé
Bellanger
a,
Frédéric
Guittard
*a,
Patrícia
Lisboa
b,
Martin
Zurn
b,
Pascal
Colpo
b,
Douglas
Gilliland
b and
François
Rossi
*b
aUniversité de Nice–Sophia Antipolis, Equipe Chimie Organique aux Interfaces, Parc Valrose, 06108, Nice Cedex 2, France. E-mail: Frederic.GUITTARD@unice.fr; Fax: (+33)492076156; Tel: (+33)492076159
bEuropean Commission, Joint Research Centre, Institute for Health and Consumer Protection, TP203, Via Fermi, 21027, Ispra, Italy. E-mail: Francois.ROSSI@jrc.ec.europa.eu; Fax: (+39)0332785787; Tel: (+39)0332785443
First published on 30th November 2011
Abstract
The immobilization of biotin on transducer surfaces is a very important step for the fabrication of biosensors for many applications (immunoassay, DNA-hybridization assays, targeted imaging). Biotinylated polypyrroles have been studied and tested but gave rise to problems of polymerization and stability due to the intrinsic properties of pyrrole. As an alternative, biotinylated pyrroles were often used in a copolymerization with pyrrole or with an amphiphilic pyrrole derivative in a copolymerization to reduce the problems due to the pyrrole substitution. To find a new strategy, this paper presents the homopolymerization, instead of the use of a copolymerization, by replacing pyrrole by 3,4-ethylenedioxypyrrole bearing biotinylated substituent. We report the synthesis, characterization and electrochemical properties of two biotinylated 3,4-ethylenedioxypyrroles differing by the length of the alkyl spacer (ethyl or dodecyl) as well as the characterization of the corresponding polymer films. We successfully show, by cyclic voltammetry, that these monomers polymerize perfectly and give relatively stable polymer films. The increase of the alkyl spacer improves the polymerization and increases the polymer stability. For the first time, we also studied the surface morphology of an electrodeposited biotinylated polymer. The electrodeposition of these biotinylated derivatives gave rise to the ability to modulate the surface microstructuration, which consists of microspheres or cauliflower-like microstructures according to the length of the alkyl spacer.
Introduction
The very high non-covalent interactions between biotin and glycoproteins such as avidin and streptavidin makes biotin an ideal candidate for many applications, for example in immunoassays, DNA-hybridization assays, targeted imaging or drug delivery.1–3 For some of these applications, biotin needs to be immobilized on surfaces for further bio-recognition for instance.Classical methods of immobilization are based on grafting or polymer deposition. Electrochemical polymerization is a very fast method for the deposition of conducting polymers using soft conditions, which can be employed, in certain conditions, with biological species such as biotin.4–19 Among all the electronically conducting polymers, polypyrrole is often used because of its biocompatibility and soft electropolymerization conditions.4–16 Usually, biotin is either incorporated inside electroactive polymers as a doping compound thanks to its carboxylic group,14 or grafted on the monomer before polymerization.4–13 The biotin doping allowed a maximum of one biotin per three–four monomer units and presented polymer stability problems or doping loss with time. To avoid this problem, biotin can be chemically grafted on the monomer. In this method, the substituent size (biotin + spacer + connector) has a very significant influence on the polymerization ability and has a consequence on polymer chain lengths, dispersion in size and solubility. In other cases, to facilitate the polymerization, biotin was grafted on the polymer using a post-treatment,4 which leads to lower concentration of biotin.
In all the publications reported in the literature on biotin-substituted pyrrole, biotin was chemically linked to pyrrole in the N-position. Many pyrrole derivatives containing biotin at the N-position and with various spacers were synthesized and analyzed by many groups, as represented in Scheme 1A. However, the polymerization of pyrrole derivatives was very sensitive to the substituent presence and in the case of pyrrole with substitution at the N-position, very high steric hindrances were induced20,21 giving rise to problems of stability as previously reported by Cosnier et al.8 To reduce steric hindrances and to make the electrodeposition easier, a biotinylated pyrrole was often mixed with pyrrole or an amphiphilic pyrrole derivative in a copolymerization process.4–10,16N-Biotinylated polypyrroles showing biological activity have been reported where the carboxylic group of biotin was not much involved in the molecular recognition.
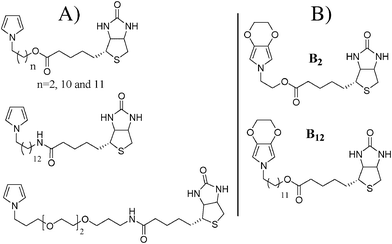 | ||
| Scheme 1 (A) Biotinylated pyrroles reported in the literature and (B) schematic representation of the biotinylated 3,4-ethylenedioxypyrroles studied in this paper. | ||
One of the methods to improve the electropolymerization would be to put the substituent in the 3-position. However, it is now established that polypyrrole films often present many surface defects such as wrinkles which are not wanted for many applications.22–24 Very recently, a new family of polypyrrole derivatives, known as poly(3,4-alkylenedioxypyrroles), showing exceptional electronic and optical properties, has been developed.25–29 In this series, the presence of the 3,4-alkylenedioxy bridge has many advantages. First of all, if the 3- and 4-position are occupied, no structural defects are possible. Moreover, the electron donating properties of the bridge highly decrease the monomer and polymer oxidation potentials rendering the electropolymerization much easier. Fluorinated poly(3,4-ethylenedioxypyrroles) (PEDOP) and poly(3,4-propylenedioxypyrroles) (ProDOP) with exceptional electrochemical properties and stability and with various surface morphologies have been reported.30–31
The aim of this work is to report the synthesis and electrochemical properties of two biotinylated EDOP monomers represented in Scheme 1B. The two monomers (B2 and B12) differ by the length of the alkyl spacer between the polymerizable core and the biotin. Their ability to form polymer films electrochemically was analyzed by cyclic voltammetry. The polymer films were analyzed by imaging infrared, optical profilometry and scanning electron microscopy (SEM) in order to determine their morphology. Because the presence of nano/microstructures is very important for the elaboration of biosensors,32–35 due to the large specific surface area, three different surfaces were chosen for the electrodeposition: smooth gold plates, microstructured gold plates and nanostructured gold plates. The evaluation of the surface structuration effect on the molecular recognition power of the biotinylated surfaces will be evaluated in a future project.
Experimental
1. Monomer synthesis and characterization
All chemical were purchased from Sigma-Aldrich. EDOP, 2-tert-butyldimethylsilyloxyethyltosylate (Tos2) and 2-(2H-[1,4]dioxino[2,3-c]pyrrol-6(3H)-yl)ethanol (OH2) were obtained using a procedure already reported in the literature (Scheme 2).30 The NMR spectra were obtained with a Bruker Avance 200 MHz. The mass spectra of the molecules were realized either by electron ionisation at 70 eV with a Thermo TRACEGC of Thermofischer Corp. fitted with an Automass III Multi spectrometer (GC-MS) or with a Thermo LCQ Classic « Ion Trap » spectrometer of Thermofischer Corp. fitted with ESI Type API.1 (ESI-SM).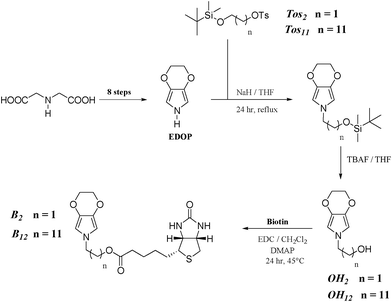 | ||
| Scheme 2 Synthetic route to the monomers. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) gave the product (retention time: 16.4 min).
:1) gave the product (retention time: 16.4 min).
Yield 54%; δH(250 MHz, CDCl3) 0.04 (6 H, s), 0.89 (9 H, s), 1.44 (20H, m) and 3.62 (4H, m).
:4) gave the product.
Yield 45%; δH(250 MHz, CDCl3) 0.04 (6 H, s), 0.88 (9 H, s), 1.39 (20 H, m), 2.44 (3 H, s), 3.59 (2 H, t, 3JHH 6.5), 4.01 (2 H, t, 3JHH 6.5), 7.34 (2 H, d, 3JHH 8.2) and 7.79 (2 H, d, 3JHH 8.2); δC(50 MHz, CDCl3) −5.27, 18.36, 21.61, 25.31, 25.78, 25.97, 28.80, 28.91, 29.36, 29.41, 29.45, 29.51, 29.58, 32.87, 63.32, 70.69, 127.87, 129.77, 133.26 and 144.57.
To this mixture, was added tetrabutylammonium fluoride in THF. After stirring during 24 h at 60 °C and solvent evaporation, the purification by column chromatography (silica gel; eluent: dichloromethane) afforded the product as crystalline solid.
Yield 38%; crystalline solid; m.p. 29.5 °C; δH(250 MHz, CDCl3) 1.25 (16 H, s), 1.56 (2 H, quint, 3JHH 6.35), 1.66 (2 H, quint, 3JHH 6.57), 3.63 (4 H, m), 4.17 (4 H, s) and 6.05 (2H, s); δC(50 MHz, CDCl3) 25.70, 26.66, 29.18, 29.38, 29.44, 29.48, 29.53, 31.30, 32.78, 50.51, 63.05, 65.83, 100.96 and 131.62; m/z 309 (M+, 1), 278 (C17H28NO2+, 1), 264 (C16H26NO2+, 2), 250 (C15H24NO2+, 3), 236 (C14H22NO2+, 4), 222 (C13H20NO2+, 6), 208 (C12H18NO2+, 6), 194 (C11H16NO2+, 11), 180 (C10H14NO2+, 15), 166 (C9H12NO2+, 6), 152 (C8H10NO2+, 14), 139 (C7H9NO2+, 100), 138 (C7H8NO2+, 61) and 125 (C6H7NO2+, 36).
(B2) Yield 65%; white solid; δH(250 MHz, CD3OD) 1.28–1.78 (6 H, m), 2.34 (2 H, t, 3JHH 7.16), 2.70 (1 H, d, 2JHH 12.70), 2.94 (1 H, dd, 2JHH 12.70, 3JHH 4.43), 3.20 (1 H, m), 3.92 (2 H, t, 3JHH 5.21), 4.10 (4 H, s), 4.23 (2 H, t, 3JHH 5.21), 4.29 (1 H, dd, 3JHH 7.87, 3JHH = 4.64), 4.50 (1 H, dd, 3JHH 7.87, 3JHH 4.43) and 6.09 (2 H, s); δC(50 MHz, CD3OD) 25.87, 29.43, 29.60, 34.78, 41.08, 56.89, 61.67, 63.40, 65.36, 66.98, 102.48, 133.54, 165.98, 175.11; νmax(film)/cm−1 3280 (NH), 2925, 2864, 1731 (COO), 1694 (NHCONH), 1551, 1391, 1362 and 1055; ESI-MS m/z 418.1 (M+Na).
(B12) Yield 59%; white solid; δH(250 MHz, CD3OD) 1.28–1.78 (26 H, m), 2.34 (2 H, t, 3JHH 7.21), 2.70 (1 H, d, 2JHH 12.70), 2.93 (1 H, dd, 2JHH 12.70, 3JHH 4.80), 3.20 (1 H, m), 3.65 (2 H, t, 3JHH 6.87), 4.10 (6 H, m), 4.30 (1 H, dd, 3JHH 7.84, 3JHH 4.40), 4.49 (1 H, dd, 3JHH 7.84, 3JHH 4.80) and 6.04 (2 H, s); δC(50 MHz, CD3OD) 25.87, 25.93, 26.00, 27.03, 27.67, 29.46, 29.68, 29.72, 30.27, 30.29, 30.58, 32.50, 34.84, 34.88, 41.02, 56.96, 61.45, 61.61, 63.38, 65.60, 66.95, 101.96, 133.04, 166.10 and 175.51; νmax(film)/cm−1 3228 (NH), 2923, 2852, 1733 (COO), 1699 (NHCONH), 1552, 1392, 1361 and 1056; ESI-MS m/z 558.3 (M+Na).
2. Elaboration of micro-patterned gold plates
The structures were micro-patterned using optical lithography based on a Microposit positive resist S1813-G2. The resist film was spin-coated onto the substrate at 3000 rpm resulting in thickness of approximately 1.5 μm. The photo resist was structured using a UV source illuminating a Digital Mirror Device with about 600000 individually switchable micro mirrors. The resulting pattern was projected onto the substrate. The illuminated part of the substrate was removed in a development step, while the unexposed part remained as a structure of mesa-shaped objects with interconnections (cf.Fig. 7). In a subsequent step the structure was metal coated.
3. Elaboration of nano-structured gold/SiOx plates using nano-template fabrication
The gold/SiOx nano-templates were fabricated by using a procedure already described elsewhere33,34 and illustrated in Fig. 1. Briefly, the fabrication consisted of the creation of a 2D hexagonal crystalline monolayer mask of self assembled polystyrene nano-beads (nominal diameter 500 nm) (Sigma-Aldrich) on the gold surface by spin coating. An oxygen plasma etching was then performed to reduce the bead diameter and to define the size of the nano-mask. Afterwards, an insulating SiOx layer was deposited through the nano-mask by Plasma Enhanced Chemical Vapour Deposition to form the nano-templates. Finally the beads were removed in water/ultrasound baths to obtain the SiOx nano-template and form gold nano-electrodes array. The mean height of the SiOx nano-template was about 14.5 nm (Fig. 2).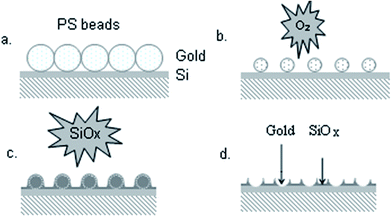 | ||
| Fig. 1 Schematic representation of the fabrication of the gold/SiOx nano-templates. | ||

4. Electrochemical polymerization
For the electrochemical experiments, 1 mM of the monomer and 0.1 M of tetrabutylammonium hexafluorophosphate (Bu4NPF6) were dissolved in anhydrous acetonitrile and degassed. A three-electrode system was used: a working electrode (platinum disk for cyclic voltammetry experiments, smooth or pattern gold plates for chronoamperometry experiments), a graphite counter-electrode and a saturated calomel reference electrode (SCE).Results and discussion
1. Synthesis
The two EDOP derivatives containing a hydroxyl group and differing by the alkyl spacer (ethyl or dodecyl) represented in Scheme 2 were obtained in ten steps from iminodiacetic acid following a synthetic route reported in previous work.30 The synthesis route includes obtaining EDOP in eight steps, the introduction of a hydroxylated substituent at the N-position using a molecule containing both a silyl and a tosylate group (2-tert-butyldimethylsilyloxyethyltosylate for B2 and 2-tert-butyldimethylsilyloxydodecyltosylate for B12). The successful synthesis of these two derivatives was confirmed by 1H and 13C NMR, infrared and mass spectrometry (cf. ESI). Afterwards, the biotinylated monomer was obtained by coupling in dichloromethane with EDC and DMAP.2. Electrochemical characterization
Each biotinylated monomer was separately introduced inside a cell containing a solution of tetrabutylammonium hexafluorophosphate (0.1 M) in anhydrous acetonitrile for the electrodeposition. A monomer concentration of 1 mM was chosen (a higher monomer concentration can lead to a decrease in the electrochemical stability). First of all, their oxidation potential, Eox = 0.91 V vs. SCE for B2 and 0.86 V for B12, was determined by potential scanning between 0 and 1.2 V. Therefore, the increase of the alkyl spacer seemed to have a beneficial effect on their oxidation. Then, their electropolymerization ability was studied by cyclic voltammetry. Their cyclic voltammograms (10 scans) are represented in Fig. 3A for B2 and 3C for B12. These graphs exhibited a reversible system, which corresponds to the oxidation and the reduction of the electroactive polymer. The increase in the intensity after each scan means that the amount of electrodeposited polymer regularly increases when the monomer oxidation potential is reached. Then, after ten scans, the working electrode was washed and put in a solution without monomer, and the stability of the films and was determined by performing 100 scans by cycling voltammetry. First of all, the half-wave potential of the polyB2 (Fig. 3B), about 0.29 V, was higher than that of non-substituted PEDOP (E1/2 = −0.24 V) and semi-fluorinated PEDOP (E1/2 = 0.24 V), whereas that of polyB12 (Fig. 3D), about 0.16 V, was much lower. As a consequence, the alkyl chain increase favoured electropolymerization, increasing the polymer chains length, while the presence of the biotin induced higher steric hindrance than semi-fluorinated tails (F-octyl) giving rise to a shortening of the polymer chains. Indeed, the biotin should be sufficiently far from the polymer backbone to not impede the intra- and intermolecular electronic exchanges. As a consequence, the cyclic voltammogram of polyB12 was an extremely well-defined doping system with three oxidation and reduction peaks, possible only if the polymer backbone is not highly disrupted,30a while that of polyB2 displayed only one oxidation and reduction peak. The polymer oxidation potentials were far from the monomer oxidation potentials, which indicated the polymer chain lengths were relatively important. Indeed, the polymers were completely insoluble in acetonitrile and no soluble part was observed during the deposition, which is a very important point in the polymer electrodeposition. Finally, the 100 scans revealed that just a slight decrease in the intensity of the polymer oxidation was detected for polyB12 (Fig. 3D) and a higher decrease for polyB2 (Fig. 3B), confirming the higher steric hindrances in this film. We also observed the very high adherence of the film on the working electrode and the difficulty to remove it after the experiments.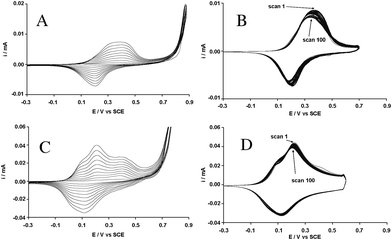 | ||
| Fig. 3 Cyclic voltammograms of (A) B2 and (C) B12 (1 mM) in 0.1M Bu4NPF6/CH3CN: 10 scans vs. SCE. Cyclic voltammograms of (B) polyB2 and (D) polyB12 in 0.1M Bu4NPF6/CH3CN without monomer: 100 scans vs. SCE. | ||
In a recent publication, the group of Reeman reported an electrochemical method to characterize the bioaffinity of biotinylated polyterthiophenes in the presence of avidin.17a They showed that the exposure of the polymers to a buffer solution containing avidin can lead to drastic changes in the cyclic voltammograms because the presence of avidin decreases the electronic exchanges. This method was used to characterize polyB2 and polyB12. More precisely, the working electrodes containing the polymers were, first, put in a buffer solution (1 M NaCl, 10 mM EDTA) during 15 mn and a scan was performed to evaluate their resistance to the buffer. Unfortunately, polyB2 was affected by the buffer confirming its lower stability while polyB12 was completely unaffected. Then, polyB12 was added to a buffer solution containing different amounts of avidin (from 10−12 to 10−8 M) and the film was characterized by cyclic voltammetry after each exposure. The cyclic voltammograms are gathered in Fig. 4. This figure shows that the cyclic voltammogram of polyB12 was not affected after these experiments. Hence, either the bioaffinity of these polymers is not very important or the presence of avidin does not affect the electronic exchange of these polymers. Other experiments will be undertaken in the near future to determine the bioaffinity of these surfaces.
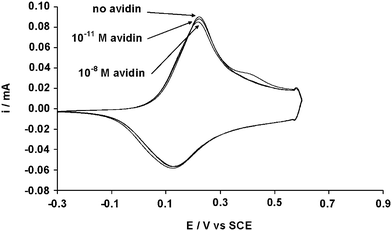 | ||
| Fig. 4 Cyclic voltammogram of polyB12 (0.01 M) after exposure to different amounts of avidin. | ||
3. Surface characterization
For the study of their surface properties, polymer films were electrodeposited on smooth and microstructured gold substrates by imposing a constant potential of 0.84 V vs. SCE for B2 and 0.80 V for B12 and with a deposition charge of 100, 200 and 300 mC cm−2. The films were characterized by infrared imaging in reflection mode. In this method, the infrared beam penetrates the polymer film and is reflected by the gold plate. Therefore, the polymer film should not be too thick or opaque. We also chose to remove the doping anions by electrochemical reduction (−0.5 V vs. SCE during 15 min) because of their absorbance in the infrared. First of all, the infrared spectra of the polymers were obtained (Fig. 5A). These spectra confirmed the presence of the polymers thanks especially to two bands: a relatively large absorption between 1680 and 1750 cm−1, due to the superposition of the ester band (1730 cm−1) and the carbamide band (1696 cm−1) of the biotin, and a large band at about 3300 cm−1 due to the N–H stretching of the carbamides. The alkyl band at 2920 and 2848 cm−1 were more intense for polyB12 due to the presence of the dodecyl spacer. Secondly, infrared imaging was used to obtain the total absorbance of the surface. According to Fig. 5B, domains where the polymer is concentrated are visualized showing that the surface seems to be structured.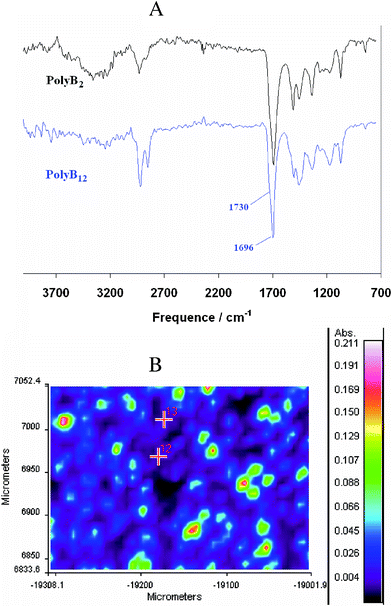 | ||
| Fig. 5 Cyclic (A) infrared spectra of dedoped polyB2 and polyB12 obtaining by imaging infrared and (B) total absorption of the de-doped polyB12 (Qs = 200 mC cm−2). | ||
The roughness of the surfaces was evaluated by optical profilometry (cf.Fig. 6 and Table 1). Table 1 gathers the roughness data of polyB2 and poly B12 as a function of the deposition charge (Qs). An increase of the surface roughness with Qs was measured for the two polymers. Electrodeposited polyB12 films were rougher, with the same polymerization conditions, than polyB2 films.
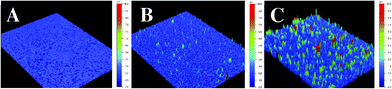 | ||
| Fig. 6 Cyclic optical profilometry images of polyB12 deposited on smooth gold plates for a deposition charge of (A) 100, (B) 200 and (C) 300 mC cm−2. | ||
| Polymer | Qs [mC cm−2] | Roughness parameters | |
|---|---|---|---|
| Ra [nm] | Rq [nm] | ||
| PolyB2 | 100 | 67.4 | 125.0 |
| 200 | 117.5 | 279.4 | |
| 300 | 556.4 | 1165.0 | |
| PolyB12 | 100 | 162.1 | 266.1 |
| 200 | 312.4 | 457.6 | |
| 300 | 893.9 | 1550.1 | |
To better evaluate the influence of the alkyl spacer on the surface roughness and morphology, the polymer films were analyzed by SEM (Fig. 7). First of all, the polymer films were very structured. The same surface morphology was observed when the polymers were electrodeposited on smooth or micro-patterned gold surfaces. The two polymer films were very structured but differences in surface morphology were observed. PolyB2 films were composed of an assembly of micrometric spherical particles of different sizes. The spherical structures were relatively smooth as shown in Fig. 7C. On the other hand, polyB12 consisted of cauliflower-like microstructures. The structures had micro and sub-micro dimensions, as shown in Fig. 7F, confirming the higher roughness of these films.
 | ||
| Fig. 7 SEM images of polyB2 (scale bar: (A) 200 μm, (B) 10 μm and (C) 3 μm) and polyB12 (scale bar : (D) 200 μm, (E) 10 μm and (C) 3 μm) electrodeposited on micro-patterned gold plates; Qs = 300 mC cm−2. | ||
The possibility of having nano-structured films of the polymer opens the possibility of many applications, as for example biosensors with increased sensitivity.35 Many trials using electrodes consisting of arrays of insulating SiOx nano-pillars showed the possibility of producing nanostructured biotinylated poly(3,4-ethylenedioxypyrrole) surfaces with a deposition charge below 5 mC per cm2 of projected area (the local density is much higher in nanostructured surfaces) (Fig. 8A). For a deposition above 5 mC cm−2, the polymer growth covered the SiOx nano-pillars as shown in Fig. 8B. Therefore, the thickness of the electrodeposited polymer is about 15 nm for a deposition charge of 5–7.5 mC cm−2.
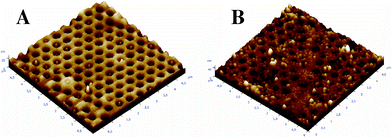 | ||
| Fig. 8 AFM pictures of polyB2 electrodeposited on nano-structured gold/SiOx plates with a deposition charge of (A) 5 and (B) 7.5 mC cm−2; the area represents 5 × 5 μm2. | ||
Conclusions
In aiming to improve the fabrication of biosensors based on the biotin-avidin system by electrodeposition, two biotinylated monomers based on the 3,4-ethylenedioxypyrrole core and differing by the alkyl spacer (ethyl or dodecyl) were synthesized, characterized and electrodeposited.First of all, the two monomers perfectly polymerized and the polymer films were very adherent, and microstructured. We showed that a long alkyl spacer (dodecyl for example) improves the polymerization and the electrochemical stability. The alkyl spacer length also has a noticeable effect on the surface morphology. The polymerization was applied to micro and nanostructured surfaces and was successfully in creating spatially controlled and structured films which may offer new opportunities for the production of novel structured sensor surfaces.
Acknowledgements
The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement no CP-FP 214478-2.References
- (a) E. P. Diamandis and T. K. Christopoulos, Clin. Chem., 1991, 37, 625 CAS; (b) M. Wilchek and E. A. Bayer, Methods Enzymol., 1990, 184, 14 CrossRef CAS.
- (a) C. M. Yam, M. Deluge, D. Tang, A. Kumar and C. Cai, J. Colloid Interface Sci., 2006, 296, 118 CrossRef CAS; (b) R. J. McMahon, Methods Mol. Biol., 2007, 418, 443 Search PubMed.
- (a) C.-M. Yam, C.-M. Pradier, M. Salmain, N. Fischer-Durand and G. Jaouen, J. Colloid Interface Sci., 2002, 245, 204 CrossRef CAS; (b) C.-M. Yam, C.-M. Pradier, M. Salmain, P. Marcus and G. Jaouen, J. Colloid Interface Sci., 2001, 235, 183 CrossRef CAS.
- S. Cosnier, Biosens. Bioelectron., 1999, 14, 443 CrossRef CAS.
- S. Cosnier, C. Gondran and A. Senillou, Synth. Met., 1999, 102, 1366 CrossRef CAS.
- L. M. Torres-Rodriguez, A. Roget, M. Billon, T. Livache and G. Bidan, Chem. Commun., 1998, 1993 RSC.
- G. Bidan, M. Billon, T. Livache, G. Mathis, A. Roget and L. M. Torres-Rodriguez, Synth. Met., 1999, 102, 1363 CrossRef CAS.
- S. Cosnier and A. Lepellec, Electrochim. Acta, 1999, 44, 1833 CrossRef CAS.
- L. M. Torres-Rodriguez, M. Billon, A. Roget and G. Bidan, Synth. Met., 1999, 102, 1328 CrossRef CAS.
- G. Bidan, M. Billon, K. Galasso, T. Livache, G. Mathis, A. Roget, L. M. Torres-Rodriguez and E. Vieil, Appl. Biochem. Biotechnol., 2000, 89, 183 CrossRef CAS.
- (a) S. Cosnier, M. Stoytcheva, A. Senillou, H. Perrot, R. P. M. Furriel and F. A. Leone, Anal. Chem., 1999, 71, 3692 CrossRef CAS; (b) R. S. Marks, A. Novoa, D. Thomassey and S. Cosnier, Anal. Bioanal. Chem., 2002, 374, 1056 CrossRef CAS.
- (a) A. Dupont-Fillard, M. Billon, T. Livache and S. Guillerez, Anal. Chim. Acta, 2004, 515, 271 CrossRef; (b) A. Dupont-Fillard, A. Roget, T. Livache and M. Billon, Anal. Chim. Acta, 2001, 449, 45 CrossRef; (c) A. Dupont-Fillard, M. Billon, S. Guillerez and G. Bidan, Talanta, 2001, 55, 981 CrossRef.
- (a) S. Cosnier, B. Galland, C. Gondran and A. Lepellec, Electroanalysis, 1998, 10, 808 CrossRef CAS; (b) R. Haddad, S. Cosnier, A. Maaref and M. Holzinger, Analyst, 2009, 134, 2412 RSC.
- P. M. George, D. A. LaVan, J. A. Burdick, C.-Y. Chen, E. Liang and R. Langer, Adv. Mater., 2006, 18, 577 CrossRef CAS.
- N. Haddour, S. Cosnier and C. Gondran, Chem. Commun., 2004, 2472 RSC.
- I. I. Nita, K. Abu-Rabeah, A. M. Tencaliec, S. Cosnier and R. S. Marks, Synth. Met., 2009, 159, 1117 CrossRef CAS.
- (a) F. Mouffouk, S. J. Brown, A. M. Demetriou, S. J. Higgins, R. J. Nichols, R. M. G. Rajapakse and S. Reeman, J. Mater. Chem., 2005, 15, 1186 RSC; (b) F. Mouffouk and S. J. Higgins, Electrochem. Commun., 2006, 8, 15 CrossRef CAS.
- S. J. Higgins, F. Mouffouk, S. J. Brown, N. Sedghi, B. Eccleston and S. Reeman, Mater. Res. Soc. Symp. Proc., 2005, 871E, I1.3.1 Search PubMed.
- F. Mouffouk, S. J. Brown, A. M. Demetriou, S. J. Higgins, R. J. Nichols, R. M. G. Rajapakse and S. J. Reeman, J. Mater. Chem., 2005, 15, 1186–1196 RSC.
- W. J. Feast, J. Tsibouklis, K. L. Pouwer, L. Groenendaal and E. W. Meijer, Polymer, 1996, 37, 5017 CrossRef CAS.
- K. K. Kanazawa, A. F. Diaz, R. H. Geiss, W. D. Gill, J. F. Kwak, J. A. Logan, J. F. Rabolt and G. B. J. Street, J. Chem. Soc., Chem. Commun., 1979, 854 RSC.
- S. J. Sutton and A. S. Vaughan, Polymer, 1995, 36, 1849 CrossRef CAS.
- J. S. Shapiro, W. T. Smith and C. MacRae, Synth. Met., 1995, 69, 505 CrossRef CAS.
- M. J. Miles, W. T. Smith and J. S. Shapiro, Polymer, 2000, 41, 3349 CrossRef CAS.
- R. M. Walczak and J. R. Reynolds, Adv. Mater., 2006, 18, 1121 CrossRef CAS.
- C. A. Thomas, K. Zong, P. Schottland and J. R. Reynolds, Adv. Mater., 2000, 12, 222 CrossRef CAS.
- P. Schottland, K. Zong, C. L. Gaupp, B. C. Thompson, C. A. Thomas, I. Giurgiu, R. Hickman, K. A. Abboud and J. R. Reynolds, Macromolecules, 2000, 33, 7051 CrossRef CAS.
- G. Sönmez, I. Schwendeman, P. Schottland, K. Zong and J. R. Reynolds, Macromolecules, 2003, 36, 639 CrossRef.
- C. L. Gaupp, K. Zong, P. Schottland, B. C. Thompson, C. A. Thomas and J. R. Reynolds, Macromolecules, 2000, 33, 1132 CrossRef CAS.
- (a) T. Darmanin and F. Guittard, J. Am. Chem. Soc., 2009, 131, 7928 CrossRef CAS; (b) T. Darmanin and F. Guittard, J. Colloid Interface Sci., 2009, 335, 146 CrossRef CAS; (c) T. Darmanin and F. Guittard, J. Mater. Chem., 2009, 19, 7130 RSC.
- (a) A. Zenerino, T. Darmanin, E. Taffin de Givenchy, S. Amigoni and F. Guittard, Langmuir, 2010, 26, 13545 CrossRef CAS; (b) T. Darmanin, E. Taffin de Givenchy, S. Amigoni and F. Guittard, Langmuir, 2010, 26, 17596 CrossRef CAS; (c) T. Darmanin, F. Guittard, S. Amigoni, E. Taffin de Givenchy, X. Noblin, R. Kofman and F. Celestini, Soft Matter, 2011, 7, 1053 RSC.
- S. W. Lee, B. Kim, D. S. Lee, J. G. Park, S. J. Ahn, E. E. B. Campbell and Y. W. Park, Nanotechnology, 2006, 17, 992 CrossRef CAS.
- (a) L. Xia, Z. Wei and M. Wan, J. Colloid Interface Sci., 2010, 341, 1 CrossRef CAS; (b) K. H. An, S. Y. Jeong, H. R. Hwang and Y. H. Lee, Adv. Mater., 2004, 16, 1005 CrossRef CAS.
- A. Valsesia, P. Lisboa, P. Colpo and F. Rossi, Anal. Chem., 2006, 78, 7588 CrossRef CAS.
- P. Lisboa, A. Valsesia, P. Colpo, F. Rossi and M. Mascini, Anal. Lett., 2010, 43, 1556 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: NMR analyses. See DOI: 10.1039/c1ra00392e/ |
| This journal is © The Royal Society of Chemistry 2012 |