The role of anisotropic structure and its aspect ratio: high-loading carbon nanospheres supported Pt nanowires with high performance toward methanol electrooxidation†
Fengzhan
Si
ac,
Liang
Ma
ac,
Changpeng
Liu
a,
Xinbo
Zhang
*b and
Wei
Xing
*a
aState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, 5625 Renmin Street, Changchun, China. E-mail: xingwei@ciac.jl.cn; Fax: 86-431-85685653; Tel: 86-431-85262223
bState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, 5625 Renmin Street, Changchun, China. E-mail: xbzhang@ciac.jl.cn; Fax: 86-431-85262235; Tel: 86-431-85262235
cGraduate University of Chinese Academy of Sciences, Beijing, China
First published on 4th November 2011
Abstract
High-loading carbon supported Pt nanowire electrocatalysts are successfully synthesized in a mild one-pot template- and surfactant-free route. The high electrocatalytic activity, Pt utilization, and long-term stability toward methanol are ascribed to the anisotropic structural properties, exposure of specified planes, and the role of support on good dispersion.
Direct methanol fuel cells (DMFCs) show a great potential as power sources for vehicles and portable electronic devices due to their high power densities, low operating temperature, and the ease in handling using a liquid fuel.1 However, for its large-scale commercialization, great improvements are urgently needed, with the top of the list being catalyst performance.2 To this end, intensive efforts have been directed toward improving the activity and stability of the Pt-based catalysts by tuning the size, shape, structure, and/or composition of the active component3 as well as its interaction with the support.4 Among them, compared to its isotropic counterpart, nanowires (NWs) possess a higher catalytic efficiency thanks to their anisotropic structures which are largely encircled by particular planes with higher catalytic activities.3d,e,5 And for a 1D nanostructure, the aspect ratio is an important parameter to decide the ratio of a continuous surface and the exposed edge planes.6 On the other hand, in order to effectively reduce the thickness of the electrode catalyst layer and thus shorten the mass transport length of the fuel, further increasing the Pt loading while retaining the high mass activity is of great importance,3a,4a,7 while this is still a challenge because high Pt loading on carbon nanospheres tends to severely aggregate.3a,7b,c Therefore, synthesis of a high-loading high-performance Pt NWs/C catalyst is of great importance toward the large-scale application of DMFCs.
Besides their use as an electrocatalyst, NWs are also broadly applied in sensing technology, optics, electronics etc.,8 and thus many physical9 and chemical5,10 strategies have been developed for its synthesis. Among them, various templates or surfactants are employed in chemical methods,10b–d,11 which inevitably complicate the synthesis procedure and also don't ensure the purity of the product even after a strict post-treatment process.10d,11a Alternatively, anisotropic growth can be achieved by deliberately introducing foreign ions to tune the supersaturation of Pt atoms, slowing down the reduction reaction, whereas the NW growth happens only when the precursor concentration is low enough,10a which has hindered its extensive application. Furthermore, the remaining ions would poison the sample surface which is vital for catalytic applications where surface active sites are urgently needed. On the other hand, most of the synthetic methods reported to date rely heavily on organic solvents, which is mainly due to the hydrophobicity of the capping agents used. However, this will be poisonous or cause pollution in large-scale production.10b–e,11 Some facile syntheses of carbon supported Pt NWs in aqueous solution without templates, surfactants or exotic ions have been reported,3d,e but the parameters of the 1D Pt nanostructure which are important for an application and catalytic mechanism study have not been discussed. Therefore, further discussion of the effect of the support on the morphology and the dispersion of Pt NWs, and confirmation that their catalytic activity is controlled by the topography and aspect ratio is of great interest.
Herein, highly-dispersed high-loaded (60 wt%) Pt NWs in situ are grown on carbon black (Vulcan XC-72R and XC-72) and are successfully obtained via a simple and mild quasi-steady-state reduction process, wherein the carbon support also plays an important role on the morphology and dispersion of the Pt NWs. Unexpectedly, the resultant catalyst is far more active (up to 322% in specific activity) for methanol oxidation than that of the commercial Pt/C catalyst.
In a typical synthesis, the Pt4+ ions were reduced using a weak reducing agent, formic acid (HCOOH), and grown in situ on Vulcan carbon nanospheres which have been well-dispersed in ultrapure water.3e Under such conditions, Pt atoms are generated significantly slowly and are accumulated according to the lowest energy principle to grow along <111> direction. The schematic steps for the preparation are shown in Fig. 1.
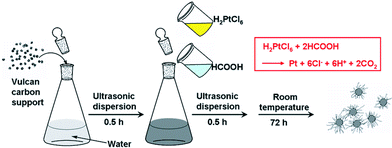 | ||
| Fig. 1 Schematic illustration of the synthesis of Pt NWs/C catalyst. | ||
Fig. 2 shows the scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of a representative sample. The SEM images with a higher resolution are in Fig. S2. †It is found that, upon adding 60 wt% of Pt, the surface of the pristine carbon spheres (Vulcan XC-72R, Fig. 2a) is thoroughly covered by a large quantity of Pt NWs which are approximately 100 nm in length (Fig. 2b), which is also confirmed by the TEM image (Fig. 2c). These densely packed arrays of Pt NWs ensure an effective high loading of Pt on the carbon support, which would accordingly improve its activity and stability (vide infra). The selected-area electron diffraction (SAED) pattern (Fig. 2c inset) shows several bright concentric rings assignable to {111}, {200}, {220}, and {311} crystal planes of fcc Pt, indicating that the obtained Pt NWs are crystallized in a phase similar to bulk Pt, which is consistent with the X-ray diffraction (XRD) result [see the ESI,† Fig. S6]. These Pt NWs/C nanostructures are further characterized by a high-resolution TEM (HRTEM) image (Fig. 2d) which shows that the Pt NWs are 3.3 nm in diameter on average. The crystallographic alignment of these NWs reveals that all of the NWs are one single crystal with a lattice spacing 0.226 nm, indicating that the NWs grow along the <111> direction. This anisotropic growth may be attributed to the very slow reduction rate at room temperature and the lowest energy principle.12
 | ||
| Fig. 2 SEM images of Vulcan XC-72R carbon nanospheres (a) before, (b) after growth of Pt NWs (60 wt%), (c) low and (d) high magnification TEM micrographs of the Pt NWs/XC-72R. The inset in (c): SAED of the Pt NWs. | ||
It should be noted that the density and length the Pt NWs on the carbon nanospheres could be controlled by the morphology of the carbon supports. Contrary to Vulcan XC-72R, when the compressed-type (Vulcan XC-72) is employed, only shorter Pt NWs (ca. 50 nm) can be obtained (Fig. S1b–d†). This might because the carbon nanospheres in Vulcan XC-72 agglomerate much more severely (Fig. S1a†), which means there is an inadequate space for the growth of the Pt NWs. These differences are expected to have a significant effect on their activity and stability. In addition, the control experiments under different reaction temperatures and times (Fig. S4 and Fig. S5†) show that dense and long Pt NWs can only be obtained at a lower temperature and after a prolonged reaction time, implying that the necessary quasi-steady-state reduction process for Pt NW growth can only be ensured at low temperature.
To compare the activity of the above described two types of Pt NWs/C and commercial Pt/C catalyst, we evaluated the electrooxidation reaction of methanol. Fig. 3 shows their linear sweep voltammetry (LSV) in deaerated 0.5 M CH3OH and 0.5 M H2SO4 solution. Unexpectedly, both the Pt NWs/XC-72R and Pt NWs/XC-72 catalysts exhibit considerably enhanced catalytic activities in terms of normalized current per unit electrochemical surface area (ECSA) (from Table S1†) of Pt (Fig. 3a–b). Specifically, the maximum current (forward scan) of the former one is more than three times (up to 322%) that of the commercial Pt/C catalyst (Fig. 3c). Considering the chemical similarity of the carbon support, the superior catalytic activity of Pt NWs/C catalysts indicates that, contrary to isotropic Pt (spherical Pt in commercial Pt/C, Fig. S3†), the anisotropic structure endows the Pt NWs with particular planes which allow a higher catalytic activity toward the electrooxidation of methanol as well as its intermediate, CO. This is further supported by the CO stripping results (Fig. S8†), showing a negative shift of the CO oxidation peak potential by about 80 mV for both Pt NW samples compared with the commercial one, demonstrating that the nano-structure is of the CO tolerance potential, which is significant for practical applications. Furthermore, compared to Pt NWs/XC-72, the much higher catalytic activity of Pt NWs/XC-72R with relatively longer Pt NWs reminds us that the aspect ratio, which was controlled by the carbon support of the Pt NWs, also plays an important role on the catalytic activity. It should be noted that, when using mass activity as the indicator of electrocatalytic quality (Fig. 3 inset), the Pt NWs/XC-72R catalyst still exhibits a greatly enhanced catalytic activity compared to the commercial one. This result further confirms the power of the anisotropic structure and the high aspect ratio of Pt NWs for electrooxidation of methanol, which might be thanks to the larger pristine surfaces with long segments of crystalline planes12 and better path-directing effects to reduce the resistance.12
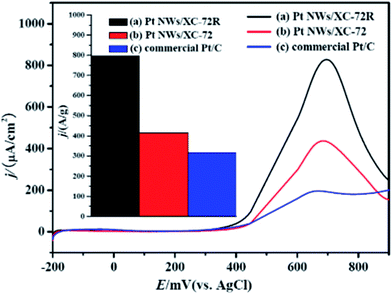 | ||
| Fig. 3 Comparison of specific catalytic activity (i.e., current per unit ECSA of catalyst) of (a) Pt NWs/XC-72R, (b) Pt NWs/XC-72, and (c) commercial Pt/C catalyst. Electrolyte: 0.5 M H2SO4 and 0.5 M CH3OH deaerated solution; sweep rate: 20 mV s−1; temperature: room temperature. Inset: comparison of the mass activity derived from Fig. S9.† | ||
The stability is of great importance for the practical application of a catalyst. In this sense, we tested the CVs for each type of the above described catalysts over 1000 cycles (Fig. S10†) and chronoamperometry curves over 4 h (Fig. S11†). It is found that there are fewer decreases in the current density (catalytic activity) with the NW-type catalysts, especially Pt NWs/XC-72R with a higher aspect ratio. This might because the 1D nanostructure possesses fewer lattice boundaries and surface defect sites when the aspect ratio is higher, which are susceptible to oxidation, decomposition,13a dissolution and subsequent Ostwald ripening.13b 1D architecture is thus considered to be a promising choice for designing a stable DMFC anodic catalyst.
In summary, we report the synthesis of Pt NWs with controlled aspect ratios on two kinds of carbon support via a facile and mild quasi-steady-state reduction approach. Surprisingly, the resultant high-loading Pt NWs/C catalyst combines excellent electrocatalytic activity, high Pt utilization, and long-term stability toward methanol oxidation, wherein the 1D nano-structure and its high aspect ratio play a key role. The obtained catalyst is believed to open new and exciting possibilities to improve the performance of DMFCs, and these encouraging findings about the power of anisotropic structure and aspect ratio could be expanded to other systems or reactions and could open up a new direction in catalysis. Besides as catalysts, it could be used for optical, sensor and electrical applications.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (20933004, 21011130027, 21073180), National Basic Research Program of China (973 Program, 2011CB935702), High Technology Research Program (863 program, 2007AA05Z159 and 2007AA05Z143) of Science and Technology Ministry of China, and Jilin Province Science and Technology Development Program (20100102).References
- (a) A. Tomita, J. Nakajima and T. Hibino, Angew. Chem., Int. Ed., 2008, 47, 1462–1464 CrossRef CAS; (b) T. Hyeon, S. Han, Y. E. Sung, K. W. Park and Y. W. Kim, Angew. Chem., Int. Ed., 2003, 42, 4352–4356 CrossRef CAS; (c) B. D. McNicol, J. Electroanal. Chem., 1981, 118, 71–87 CAS; (d) T. B. Reed and R. M. Lerner, Science, 1973, 182, 1299–1304 CAS; (e) B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS; (f) J. M. Tour, C. Kittrell and V. L. Colvin, Nat. Mater., 2010, 9, 871–874 CrossRef CAS; (g) C. Y. Wang, Chem. Rev., 2004, 104, 4727–4765 CrossRef CAS.
- (a) J. Kim, T. Momma and T. J. Osaka, J. Power Sources, 2009, 189, 999–1002 CrossRef CAS; (b) Z. B. Wang, G. P. Yin and P. F. Shi, J. Power Sources, 2007, 163, 688–694 CrossRef CAS; (c) A. D. Anderson, G. A. Deluga, J. T. Moore, M. J. Vergne, D. M. Hercules, E. A. Kenik and C. M. Lukehart, J. Nanosci. Nanotechnol., 2004, 4, 809–816 CrossRef CAS.
- (a) B. Fang, N. K. Chaudhari, M. S. Kim, J. H. Kim and J. S. Yu, J. Am. Chem. Soc., 2009, 131, 15330–15338 CrossRef CAS; (b) X. Y. Fu, Y. A. Wang, N. Z. Wu, L. L. Gui and Y. Q. Tang, Langmuir, 2002, 18, 4619–4624 CrossRef CAS; (c) L. Ma, C. P. Liu, J. H. Liao, T. H. Lu, W. Xing and J. J. Zhang, Electrochim. Acta, 2009, 54, 7274–7279 CrossRef CAS; (d) S. H. Sun, F. Jaouen and J. P. Dodelet, Adv. Mater., 2008, 20, 3900–3904 CrossRef CAS; (e) S. H. Sun, D. Q. Yang, D. Villers, G. X. Zhang, E. Sacher and J. P. Dodelet, Adv. Mater., 2008, 20, 571–574 CrossRef CAS.
- (a) S. Y. Huang, P. Ganesan, S. Park and B. N. Popov, J. Am. Chem. Soc., 2009, 131, 13898–13899 CrossRef CAS; (b) M. Okamoto, T. Fujigaya and N. Nakashima, Small, 2009, 5, 735–740 CrossRef CAS; (c) C. Wang, M. Waje, X. Wang, J. M. Tang, R. C. Haddon and Y. S. Yan, Nano Lett., 2004, 4, 345–348 CrossRef CAS.
- C. Koenigsmann, W. P. Zhou, R. R. Adzic, E. Sutter and S. S. Wong, Nano Lett., 2010, 10, 2806–2811 CrossRef CAS.
- (a) D. J. Shirale, M. A. Bangar, W. Chen, N. V. Myung and A. J. Mulchandani, J. Phys. Chem. C, 2010, 114, 13375 CrossRef CAS; (b) M. S. Sander, A. L. Prieto, R. Gronsky, T. Sands and A. M. Stacy, Adv. Mater., 2002, 14, 665 CrossRef CAS.
- (a) G. Wu, D. Y. Li, C. S. Dai, D. L. Wang and N. Li, Langmuir, 2008, 24, 3566–3575 CrossRef CAS; (b) J. Y. Cao, C. Du, S. C. C. Wang, P. Mercier, X. G. Zhang, H. Yang and D. L. Akins, Electrochem. Commun., 2007, 9, 735–740 CAS; (c) I. S. Park, K. W. Park, J. H. Choi, C. R. Park and Y. E.Sung, Carbon, 2007, 45, 28–33 CrossRef CAS.
- (a) B. D. Gates, Nat. Nanotechnol., 2010, 5, 484–485 CrossRef CAS; (b) A. I. Hochbaum and P. D. Yang, Chem. Rev., 2010, 110, 527–546 CrossRef CAS; (c) V. Schmidt, J. V. Wittemann and U. Gosele, Chem. Rev., 2010, 110, 361–388 CrossRef CAS.
- (a) H. Y. Shi, B. Hu, X. C. Yu, R. L. Zhao, X. F. Ren, S. L. Liu, J. W. Liu, M. Feng, A. W. Xu and S. H. Yu, Adv. Funct. Mater., 2010, 20, 958–964 CrossRef CAS; (b) Z. J. Zhang, C. H. Xu and Y. Yue, Appl. Phys. Lett., 2006, 88, 133102 CrossRef; (c) S. Sen, U. M. Bhatta, V. Kumar, K. P. Muthe, S. Bhattacharya, S. K. Gupta and J. V.Yakhmi, Cryst. Growth Des., 2008, 8, 238–242 CrossRef CAS; (d) Y. Deng, Y. Xiang and Y. Z. Song, Cryst. Growth Des., 2009, 9, 3079–3082 CrossRef CAS.
- (a) J. Y. Chen, T. Herricks, M. Geissler and Y. N. Xia, J. Am. Chem. Soc., 2004, 126, 10854–10855 CrossRef CAS; (b) Y. J. Han, J. M. Kim and G. D. Stucky, Chem. Mater., 2000, 12, 2068–2069 CrossRef CAS; (c) T. Kijima, T. Yoshimura, M. Uota, T. Ikeda, D. Fujikawa, S. Mouri and S. Uoyama, Angew. Chem., Int. Ed., 2004, 43, 228–232 CrossRef CAS; (d) Y. Sakamoto, A. Fukuoka, T. Higuchi, N. Shimomura, S. Inagaki and M. Ichikawa, J. Phys. Chem. B, 2004, 108, 853–858 CrossRef CAS; (e) Z. R. Shen, M. Yamada and M. Miyake, Chem. Commun., 2007, 245–247 RSC.
- (a) Y. S. Jung, J. H. Lee, J. Y. Lee and C. A. Ross, Nano Lett., 2010, 10, 3722–3726 CrossRef CAS; (b) B. Mayers, X. C. Jiang, D. Sunderland, B. Cattle and Y. N. Xia, J. Am. Chem. Soc., 2003, 125, 13364–13365 CrossRef CAS.
- J. D. Hoefelmeyer, K. Niesz, G. A. Somorjai and T. D. Tilley, Nano Lett., 2005, 5, 435–438 CrossRef CAS.
- (a) Y. Shao, G. Yin and Y. Gao, J. Power Sources, 2007, 171, 558–566 CrossRef CAS; (b) Z. Chen, M. Waje, W. Li and Y. Yan, Angew. Chem., Int. Ed., 2007, 46, 4060–4063 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Detailed synthesis conditions; experimental procedures; electron microscope characterization of Vulcan XC-72 sample synthesized under different conditions; XRD characterization; CV in H2SO4 solution; and CO stripping experiments; tests for Pt mass activities of the samples; the cycle stability tests; CA curves. See DOI: 10.1039/c1ra00326g/ |
| This journal is © The Royal Society of Chemistry 2012 |