Making polymeric nanoparticles stimuli-responsive with dynamic covalent bonds
Alexander W.
Jackson
and
David A.
Fulton
*
Chemical Nanoscience Laboratory, School of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: d.a.fulton@ncl.ac.uk; Tel: +44 (0)191 222 7065
First published on 12th October 2012
Abstract
This review article highlights recent progress in the development of new polymeric nanoparticles which display stimuli-responsiveness on account of their incorporation of dynamic covalent bonds (DCBs). These bonds can form and cleave in response to changes in environmental parameters, such as pH or presence of reducing/oxidizing agents, and chemists have utilized this reversibility to make increasingly sophisticated stimuli-responsive polymeric nanoparticles. Another key feature of DCBs is their capacity to undergo component exchange processes involving the exchange for one reaction partner for another (for example, the reaction of an imine with an amine to produce a different imine and amine), which can present polymeric nanoparticles opportunities to modify their constitutions by reshuffling, incorporating or releasing components. The utilization of the responsive nature of DCBs presents an alternative approach towards stimuli-responsive polymeric nanoparticles which does not rely upon the utilization of ‘conventional’ stimuli-responsive polymers such as poly(N-isopropylpolyacrylamide). These new stimuli-responsive polymeric nanoparticles could find application in contemporary fields such as drug delivery or diagnostics, or within novel products in the established fields of paints, coatings and adhesives.
 Alexander W. Jackson | Alexander W Jackson grew up in the small coastal town of Whitley Bay, not far from Newcastle, and completed his MChem at Newcastle University in 2008. His Masters research project was spent in the labs of Prof Michael North studying the insertion of waste carbon dioxide into terminal epoxides to form useful cyclic carbonates, utilizing a new class of catalysts based on dimeric aluminium salen complexes. Alex became the first graduate student in the group of David Fulton in October 2008, where he developed the utilization of dynamic covalent bonds within polymeric nanoparticles, co-authoring eight peer-reviewed publications in the process. In September 2012 after completing his PhD he moved to Singapore to take up a position as a research scientist at A* STAR with the Institute of Chemical and Engineering Sciences (ICES) where he works on the development functional and biocompatible polymeric nanoparticles for targeted PET imaging. |
 David A. Fulton | David A Fulton is a Lecturer in the School of Chemistry at Newcastle University. A native of the town of Kilbirnie in north Ayrshire, Scotland, he received his BSc (Hons) from Strathclyde University in 1996 and PhD in 2001 from the University of California, Los Angeles under the direction of Prof Sir J Fraser Stoddart FRS, working on carbohydrate and supramolecular chemistry. After a brief spell in industry he then spent two and half years as a postdoctoral research associate with Prof David Parker FRS at the University of Durham working on the synthesis of gadolinium-centered dendrimers as new MRI contrast agents. In 2006 he moved up the road to take up his present position within the School of Chemistry and as a member of its Chemical Nanoscience group. His research interests are focused on using synthetic polymer chemistry to address problems in medicine, nanoscience and materials science. |
Introduction
Polymeric nanoparticles have long been utilized as key components in established everyday products such as paints, coatings and adhesives.1 More recently, polymeric nanoparticles are also starting to find application in biomedical fields, where they hold promise in applications such as drug delivery, diagnostics and bioimaging.2 For both established and up-and-coming applications, there is drive to develop new polymeric nanoparticles which possess the virtues of stimuli-responsiveness, where a nanoparticle can respond to the application of a stimulus by changing its structure, and hence its physical properties, in a way which ultimately enhances the utility of the nanoparticle. Such advances would make polymeric nanoparticles better placed to overcome some of the significant challenges in biomedical applications3 or allow the development of new everyday products displaying responsive properties.When considering how to introduce stimuli-responsiveness into polymeric nanoparticles, chemists have usually relied heavily upon well-known polymers which possess an inherent stimuli-responsiveness. These polymers can respond to input stimuli—in particular to changes in temperature or pH—by undergoing a reversible phase transition.4 Arguably the most utilized polymer here is the thermoresponsive polymer poly(N-isopropyl)acrylamide (Fig. 1a), which is readily soluble in water at room temperature but precipitates reversibly from solution when raised above its lower critical solution temperature (LCST), changing from an extended chain conformation below this temperature into a collapsed chain above this temperature.5 This feature presents what is in effect a way to switch a polymer from hydrophilic to hydrophobic simply by changing temperature, and chemists have extensively exploited this feature to make stimuli-responsive polymeric nanoparticles based upon diblock copolymer micelles. Diblock copolymers which incorporate a thermoresponsive block such as poly(N-isopropylacrylamide)-b-poly(ethylene glycol),6 are able to switch reversibly between hydrophilic-b-hydrophilic and hydrophobic-b-hydrophilic upon heating, a trigger which can be used to facilitate the non-covalent aggregation of multiple polymer chains into micelles7 (Fig. 1b), nano-sized assemblies of polymer chains which to all intents and purposes can be considered as polymeric nanoparticles. The disassembly of these micelles can be triggered by cooling, as their constituent polymer chains loose their amphiphilicity and return to their hydrophilic-b-hydrophilic state. This thermally-induced change in polymer solubility has been used extensively to facilitate reversible micellization of a wide range of diblock copolymers,8 and similar design principles can be applied to pH-responsive polymers to prepare pH-responsive micelles. Micellar systems which incorporate thermo- or pH-responsive characteristics have been shown to release encapsulated cargos of small hydrophobic molecules upon micelle disassembly triggered by changes in temperature,9 and have been extensively studied as potential vehicles for the controlled release pharmaceuticals.10 Although very useful in making thermo- and pH-responsive micelle-based nanoparticles, ‘conventional’ stimuli-responsive polymers still present a rather limited palette of structure, property and responsiveness, and chemists have been actively researching alternative means to endow polymers with the virtues of stimuli-responsiveness.
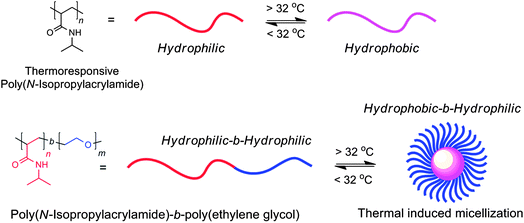 | ||
| Fig. 1 (a) Poly(N-isopropyl)acrylamide changes from an extended chain conformation below its LCST (about 32 °C) into a collapsed chain above this temperature. (b) The thermally induced reversible micellization of poly(N-isopropylacrylamide)-b-poly(ethylene glycol).6 | ||
Dynamic Covalent Bonds (DCBs)
An approach of increasing importance towards stimuli-responsive polymeric nanoparticles is the utilization of so-called dynamic covalent bonds11 (DCBs), which when judiciously placed either into or between polymer chains (Fig. 2) present key features which can be exploited to endow polymeric nanoparticles with responsive properties. The use of the term ‘dynamic’ to describe what many chemists would simply otherwise refer to as ‘reversible’ covalent bonds, was coined by the supramolecular chemistry community with its focus on studying and exploiting the dynamic nature of non-covalent bonds to prepare larger superstructures.12 The inherent chemical instability of non-covalent interactions, however, makes the resulting superstructures themselves unstable, and this lack of robustness prompted a resurgence of interest in applying chemically more stable reversible covalent bonds to prepare such molecules. The term ‘dynamic covalent bond’ describes any covalent chemical bond which possesses the capacity to be formed and broken under equilibrium control.11 It should be noted that DCBs have long played a part in polymer chemistry, where some types of polymerizations are performed under equilibrium control e.g. the formation of polyesters. It is perhaps surprising, however, that it has only been in the last decade or so that DCBs have become extensively employed to endow polymeric materials with responsive characteristics. Since the pioneering work13 by the Lehn group, the use of DCBs in polymer chemistry has seen an explosion of interest by many academic groups around the world, witnessing the development of many new dynamic covalent polymers and polymeric materials. Much of this progress has been recently reviewed,14 and the reader is strongly recommended to visit this literature should they desire to further their knowledge of DCBs in other areas of polymer chemistry beyond the nanoparticle focus of this review.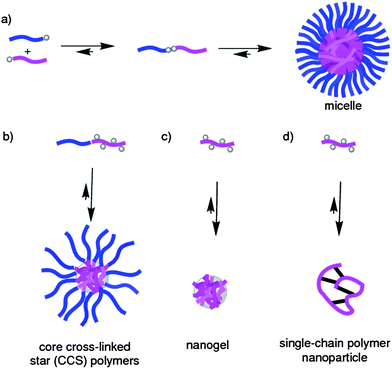 | ||
| Fig. 2 Cartoons showing some of the different classes of polymeric nanoparticles which have been endowed with stimuli-responsive properties on account of their incorporation of DCBs. (a) Two different polymer blocks can be linked through a single DCB to form an amphiphilic diblock copolymer, which can then self-assemble into a micelle. (b) Diblock copolymers possessing appropriate functional groups within one of the blocks can be cross-linked through DCBs into core cross-linked star (CCS) polymers. (c) Copolymer chains possessing appropriate functional groups can be cross-linked through DCBs into nanogel-type particles. (d) A single copolymer chain can be intramolecularly cross-linked through DCBs. | ||
The important features of DCBs which make them of such potential are best highlighted by considering, as an example, a well-known and exploited DCB: the imine bond.15 Imines are made (Fig. 3) from the condensation of an amine with an aldehyde or ketone, also producing a molecule of water in the process. This is an equilibrium process, and the position of the equilibrium is thus sensitive to environmental parameters including pH, temperature, and concentration of water. Lehn and co-workers have performed16 an in-depth investigation of the pH-sensitivity of the imine bond, one outcome of which shows that the position of the imine equilibrium can be shifted from almost complete imine to starting materials over about three pH units. By tuning the electronics of the reaction partners, it is possible to control the precise pH at which these processes occur through careful choice of amine and aldehyde. This capacity to modulate the position of equilibrium of a DCB presents the opportunity to form and cleave bonds in response to environmental parameters such as pH or presence of reducing/oxidizing agents, and chemists have made good progress in utilizing the reversibility of DCBs to make increasingly sophisticated stimuli-responsive polymeric species.
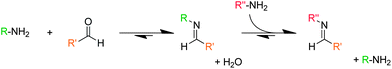 | ||
| Fig. 3 The features of DCBs which make them of such interest in polymer chemistry are highlighted using the imine bond as an example. The condensation of an amine with a carbonyl (in this instance an aldehyde) to form an imine bond and a molecule of water. The position of this equilibrium is sensitive to environmental conditions such as temperature, concentration, pH, etc. DCBs can also undergo component exchange process, illustrated here with the reaction of the imine bond with another amine to afford a new imine bond. Exchange processes can allow polymers to reshuffle or incorporate/deincorporate their components, changing their structures and thus their properties and function. | ||
Another important feature of DCBs is their capacity to undergo component exchange processes involving the exchange for one reaction partner for another, which can be illustrated (Fig. 3) by again using the imine bond as an example. The imine bond can react with an amine to produce a different imine and amine (transimination), and this component exchange feature can present polymeric species opportunities to modify their constitutions by reshuffling, incorporating or releasing components, and thus changing their structures, properties and functions.
DCBs possess further important features which makes them appealing in polymeric nanoparticles. Dynamic covalent reactions are usually performed with the help of a suitable catalyst to aid kinetics, and so the option exists to halt these reversible processes and kinetically ‘fix’ the products simply by quenching the catalyst, an option which is not available within supramolecular systems where dynamic processes cannot be easily ‘turned-off’. Hydrazone bond formation, for example, displays optimal kinetics at pH 4.5,17 and raising the pH to 7.0 can drastically slow down the rate of reaction to such an extent that it essentially becomes ‘fixed’. Alternatively, DCBs can also be ‘fixed’ through a chemical process, for example, through reduction of the imine bond with hydride reducing agents to afford the secondary amine. Another hugely important feature of DCBs is their chemical robustness, which is an important point when considering possible applications where the resulting polymeric species must possess the required levels of robustness to operate in ‘real’ environments.
There exist an extensive range of DCBs, and some of the more commonly used DCBs utilized in the field of stimuli-responsive polymeric nanoparticles are highlighted in Fig. 4. To find out more about DCBs, the reader is recommended strongly to consult the rather excellent reviews by Rowan et al.11 and Sanders et al.,18 both of which have helped to define the fields of dynamic covalent chemistry.
![Examples of DCBs commonly used in to endow polymeric nanoparticles with the virtues of stimuli-responsiveness. (a) Hydrazone formation, (b) oxime formation, (c) disulfide formation, (d) acetal/ketal formation, (e) boronic ester formation, (f) thermally induced radical cross-over reactions, and (g) Diels–Alder [4 + 2] cycloadditions.](/image/article/2013/PY/c2py20727c/c2py20727c-f4.gif) | ||
| Fig. 4 Examples of DCBs commonly used in to endow polymeric nanoparticles with the virtues of stimuli-responsiveness. (a) Hydrazone formation, (b) oxime formation, (c) disulfide formation, (d) acetal/ketal formation, (e) boronic ester formation, (f) thermally induced radical cross-over reactions, and (g) Diels–Alder [4 + 2] cycloadditions. | ||
Rather than being a comprehensive catalogue of work, in this review article we will focus on some highlights which we believe best demonstrate how polymer chemists have started to harness the appealing features of DCBs to make stimuli-responsive polymeric nanoparticles. We apologise in advance to those researchers whose work we have left out for reasons of brevity. It is hoped that this review will provide the reader with a flavour of the creativity and imagination within the state-of-the-art, and some future directions and challenges.
Micelles
A tried-and-tested methodology for making polymeric nanoparticles is the micellization of amphiphilic diblock copolymers.7 Normally these diblock copolymers possess a hydrophobic-b-hydrophilic structure with their self-assembly into micelles occurring in water, but wholly organic systems are also well-known.19 A major limitation of micelles is their inherent instability, as at low concentrations (below their critical micelle concentration) they can spontaneously disassemble into their component polymer chains. This feature of polymer micelles is a major weakness in applications such as drug delivery, where dilution effects as the nanoparticle passes through the body could lead to unwanted “burst” release of the encapsulated active compounds. To address this stability issue, cross-linking strategies have been developed in which the micellar cores20 or shells21 are covalently cross-linked, kinetically fixing the micelles. This cross-linking, however, can hinder drug release and make it harder for the body to clear the polymeric drug carriers. Consequently, chemists have been actively investigating the potential of using DCBs as cross-links which can be cleaved upon application of a desired stimulus, triggering nanoparticle disassembly and release of the encapsulated cargos.The McCormick group have reported (Fig. 5) a particularly interesting recent example which neatly highlights these concepts.22 The polymeric building block used was a hydrophilic triblock copolymer (1) comprising of poly(ethylene glycol)-b-poly(3-aminopropyl methacrylamide)-b-poly(N-isopropylacrylamide) (PEG-b-PAPMA-b-PNIPAm), which was prepared via RAFT polymerization. This triblock copolymer was shown to form micellar assemblies (2) possessing a core–shell–corona architecture above the lower critical solution temperature of the PNIPAm block. The hydrophobic nature of the cores of these micelles means they can encapsulate the hydrophobic anti-inflammatory prednisolone 21-acetate (PA), which has been used as a model drug in this study. To facilitate the cross-linking of the polymer chains, the dialdehyde terephthaldicarboxaldehyde was used to cross-link the shell blocks through imine bond formation with the amine functions of the poly(3-aminopropyl methacrylamide) blocks. The resulting nanoparticles possess both pH- and thermoresponsive properties, and the group have shown how these can be harnessed to control the release of the encapsulated drug. When the temperature is lowered to 25 °C the core becomes hydrophilic, triggering the relatively slow release of the PA cargo. Complete disassembly of the micelle is achieved through the adjustment of the pH to 5.5 at 25 °C, triggering imine hydrolysis and the fast “burst” release of PA. The ability to use different stimuli to control the rate of drug release is potentially very powerful, and this study provides much encouragement for the further development of stimuli-responsive polymeric nanoparticles in drug delivery.
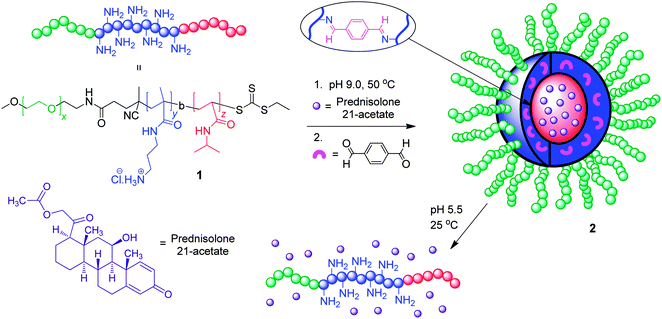 | ||
| Fig. 5 Imine shell cross-linked micellar assemblies possessing pH- and thermoresponsive characteristics.22 | ||
Another interesting application of DCBs in polymeric micelles is the concept that two polymer blocks can be linked through a single DCB to form an amphiphilic diblock copolymer (Fig. 2a). These diblock copolymers should be able to self-assemble into micelles in the same way as ‘conventional’ diblock copolymers, with the advantage that the DCB allows the incorporation of the dynamic element of the DCB into the resultant micellar nanoparticle. This concept has been investigated, initially on the smaller scale of surfactant micelles which although not polymeric in nature are still worthwhile highlighting. The group of van Esch have exploited23 the pH-responsiveness of the imine bond to prepare surfactant micelles whose disassembly/reassembly can be triggered by changes in pH. Condensation of the water-soluble aromatic aldehyde (3) (Fig. 6) with aliphatic amines (4) at pH 11.8 forms the imine amphiphile (5), which spontaneously self-assembles into micelles (6) possessing hydrodynamic radii of ∼5 nm. These micelles can complex a cargo of Nile Red, a fluorescent hydrophobic small-molecule dye which makes a useful model cargo. When the pH of the solution was lowered to 7.2, the imine linkages were hydrolysed causing the micelles to dissociate. This process is completely reversible, and when the pH was changed back to alkaline the micelles reform. In addition to pH, the imine bond is also sensitive to changes in temperature which can also be used to trigger micelle assembly/disassembly. When the temperature of the micelle solution was raised to 55 °C, the equilibrium constant of the imine bond drops to a sufficiently low value to trigger cleavage of the amphiphile and subsequent micelle disassembly. This work nicely illustrates how the pH- and temperature-responsiveness of the imine bond can be exploited, and represents a simple and potentially low-cost approach to a stimuli-responsive delivery system, but its relatively small size may prohibit its further development in some fields.
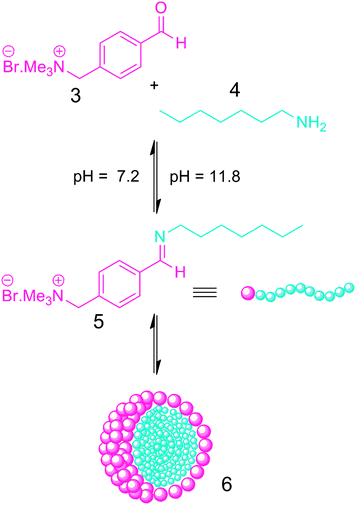 | ||
| Fig. 6 The formation of a dynamic covalent surfactant which self-assembles into surfactant micelles.23 | ||
The group of Zhang have extended24 this concept to prepare dynamic covalent bolaform amphiphiles, where the components are linked through imine bonds. Bolaform amphiphiles usually possess relatively lower critical micelle concentrations than ordinary micelles and are thus of potentially higher utility. The group have shown that micelles possessing diameters of about 18 nm can be formed which encapsulate Nile Red as a model hydrophobic cargo. As expected, a drop in pH causes hydrolysis of the imine bonds, cleavage of the bolaform amphiphile and subsequent release of the encapsulated cargo as the micelle disassembles, and the group have shown that the rate of release is pH-dependant, being fastest at low pH and slower at high pH. Like the system of van Esch, this in not a polymer-based micelle, but is also worthy of mention as the concepts could be readily ‘scaled-up’ by using polymeric building blocks.
By transposing the simple idea of an amphiphile where the two components are linked through a DCB into the realm of true polymeric nanoparticles, it should be possible to prepare larger structures possessing bigger loading capacities. In our own work we linked25 a polystyrene block with a polyisoprene block through a single dynamic covalent oxime bond, and found that the resultant dynamic covalent diblock copolymer could self-assemble into micellar nanoparticles in DMF. We showed that addition of an excess of small molecule alkoxyamine could undergo component exchange with the oxime bond, resulting in scission of the diblock copolymer and subsequent micelle disassembly. The groups of He and Zhu have looked26 at preparing polymeric micelles from diblock copolymers where a PEG block is joined to a polystyrene block through a dynamic covalent acyl hydrazone bond, and the resultant diblock copolymers was shown to form a range of morphologies in dilute DMF/water solutions. In water at pH 7 it was shown that stable micelles can be formed which are able to complex a cargo of the hydrophobe methyl porphyrin. Upon lowering the pH to 4, the disassembly of the micelles was triggered leading to the gradual release of the cargo. This work demonstrates an interesting exploitation of acylhydrazone bonds, which are normally rather stable at mildly acidic pH values.18 Presumably in this instance the choice of an electron-rich aromatic aldehyde component as a reaction partner helps to increase the susceptibility of the resulting acylhydrazone bond to acid-catalysed hydrolysis.
As previously mentioned, the study of Lehn and co-workers16 into structure-stability correlations of the imine bond in aqueous solution suggests that shifting the equilibrium of imine bond from the side of starting materials to products will occur over about three pH units. Many applications would benefit from a sharper ‘on/off’ effect over a far narrower pH value, and the group of Zhang have described27 (Fig. 7a) a ‘superamphiphile’ system which goes some way to addressing this limitation. The key to this system is its exploitation of the fact that small changes in the hydrophilic–hydrophobic balance of an amphiphilic diblock copolymer can have a very significant effect on the stability of its resultant micelle. The group have prepared pH-responsive superamphiphiles by conjugating multiple hydrophobic aldehydes (7) onto a doubly hydrophilic PEG-polylysine diblock copolymer (8). At pH 7.4 the imine equilibrium is sufficiently high such that all of the lysine amines have reacted to form a toothbrush-type amphiphile (9), essentially switching the polylysine block from hydrophilic to hydrophobic. These amphiphiles self-assemble into micelles (10) possessing hydrodynamic diameters of ∼70 nm, which were shown to complex the hydrophobic dye Nile Red. At pH 6.5 the imine bonds are sufficiently hydrolysed to remove the amphiphilicity of the component polymer building blocks, and the micelle consequently disassembles whilst releasing the Nile Red cargo. One would expect the aromatic imine bond utilized in this work not to be completely hydrolysed at pH 6.5, but it is likely that because of the poor solubility of the aldehyde component, it precipitates from solution as the imine bond is hydrolysed. This precipitation probably helps push the equilibrium further in the direction of hydrolysis. A further important factor is that as the micelle destabilizes, the imine bond becomes more unstable and is hydrolysed further. The important feature of this work is that the micelle system completely disassembles over a narrower pH range, and thus displays a sharper “on–off” behaviour. Further development of this system could lead to improved sharpening of the switching behaviour, and it should also be possible to tune the ‘on–off’ pH-switch to be matched to a desired application by altering the electronics of the aromatic aldehyde residues.
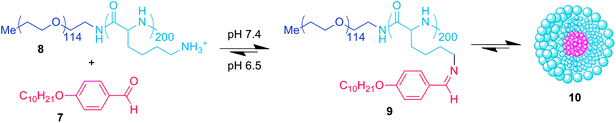 | ||
| Fig. 7 Multiple hydrophobic aldehyde units (7) conjugate onto a polylysine diblock copolymer (8) to prepare a ‘superamphiphile’ (9) which spontaneously self-assembles into a micelle (10). Lowering the pH to 6.5 causes partial hydrolysis of the imine linkages, reducing the amphiphilicity of the diblock copolymers and triggering micelle disassembly.27 | ||
Polymeric micelles are very popular candidates in drug delivery research programmes,28 and the work presented here would very much suggest that the exploitation of the DCB to endow micelles with responsive behaviours will further enhance the usefulness of micelles in this field.
Nanogels
The polymer nanogel architecture has several features which make it attractive in the field of drug delivery, with their porous architectures in particular allowing high levels of encapsulation and scope for tuneable kinetics of release. The incorporation of DCBs as cross-links into nanogels allows the introduction of stimuli-responsiveness, enhancing considerably their potential in delivery applications. The research group of Thayumanavan have lead the way in utilizing the redox-responsiveness of the disulfide bond to develop responsive nanogels for possible delivery applications.29 Although nanogels are easily prepared using emulsion polymerization techniques, it is not straightforward to introduce DCBs using this approach. Instead, the US group have developed a sophisticated polymeric system based upon linear copolymer chains (11) (Fig. 8) possessing hydrophilic (oligoethylene glycol methacrylate) and hydrophobic (2-pyridyldisulfide ethylmethacrylate) monomer units, which were shown to non-covalently aggregate into nano-sized polymeric assemblies (12) upon heating in aqueous media as a consequence of the thermoresponsive nature of the oligoethylene glycol methacrylate monomer units. The addition of a deficient amount of dithiothreitol (DTT) reduces a portion of the pyridyldisulfide functions present upon the polymer chains to alkylthiols, which then intermolecularly cross-link with the remaining pyridyldisulfide functions. The resulting cross-linked nanogel assemblies (13) were shown to be very stable at lower temperatures on account of the covalent cross-links, and were shown to non-covalently encapsulate Nile Red. The triggered release of the encapsulated Nile Red cargo is achieved through exposure to the biologically-active reducing agent glutathione, which cleaves the disulfide cross-links and triggers the disassembly of the nanogel into water-soluble linear polymer chains. The Thayumanavan group have demonstrated the capability to tune the rates of Nile Red release, adjust the nanoparticle size and adorn the nanoparticle periphery through post-formation functionalisation. It was shown that the rate of cargo release is dependent upon the concentration of glutathione, and the kinetics of cargo release can therefore be tuned by adjusting the density of disulfide cross-links, either by altering the amount of 2-pyridyldisulfide ethyl methacrylate in the linear polymer building block or by adjusting the amount of DTT added. The hydrodynamic radius of the cross-linked nanoparticles can be tuned from 10–200 nm through altering the ratio of the hydrophilic and hydrophobic monomer units in the linear polymer building blocks, or by altering the overall length of the polymer building blocks. Particle size is very important in the field of drug delivery, as it plays a crucial role in cell permeability and retention.30 Excess pyridyldisulfide functions not employed in cross-linking can be used as handles for the post-functionalisation of the nanogel assemblies, which to demonstrate this possibility were adorned with thiol-modified peptides. The potential to perform post-functionalisation is very important as biomolecules conjugated onto the nanoparticle periphery may increase specificity in cell targeting. This example highlights the chemical robustness and redox-responsive nature of nanoparticles which possess dynamic covalent disulfide cross-links.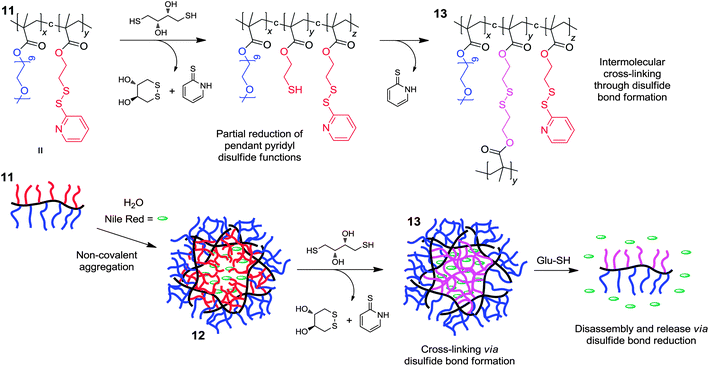 | ||
| Fig. 8 Formation of disulfide cross-linked nanogels and subsequent disassembly through disulfide bond reduction in the presence of glutathione.29 | ||
Disulfide bond formation is currently intensively investigated in the preparation of polymeric drug delivery vehicles, as the intracellular environment is known to be highly reductive.31 The reducing agent glutathione has an intracellular concentration of 3 mM, which is 300 times greater than its concentration in blood (approximately 10 μM). It is therefore widely hypothesized that polymeric nanoparticles possessing disulfide cross-links will only disassemble and release their encapsulated cargo within the intracellular environment, ensuring that drugs are only released inside of cells where they are required and not in the extracellular environment. In this regard, recent work by the group of Shuai utilizing a dual-responsive pH-responsive disulfide-cross-linked micelle to deliver the anti-cancer drug doxorubicin is noteworthy.32 This delivery system is designed to encourage burst release of the drug cargo within the lysosomes, intracelluar compartments formed during the endocytotic processes through which macromolecules often enter cells. Lysosomes possess reduced pH and high levels of glutathione, and the micellar nanoparticle developed by Shai and coworkers has been shown to deliver rapidly a drug cargo specifically within this unique environment, and thus displays much promise in delivery applications.
A limitation of many stimuli-responsive nanoparticles is their reliance upon a single stimulus to trigger a response. By requiring the simultaneous application of two different stimuli, more control is gained over where and when a particular response is triggered, and thus there has been an impetus to develop such polymeric nanoparticle systems. Our own group have recently reported33 a polymeric nanoparticle which requires the simultaneous application of two different stimuli to trigger its disassembly into its component polymer chains. This system exploits the responsiveness of two orthogonal DCBs: the pH-responsiveness of the imine bond, and the redox-responsiveness of the disulfide bond. Our nanoparticles (Fig. 9) were prepared by cross-linking multiple polymer chains through both imine and disulfide bonds to form nanoparticles possessing hydrodynamic radii of ∼80 nm. Application of either low pH or a reducing agent did not trigger the disassembly of the nanoparticle into their component polymer chains as there remains a sufficient density of either disulfide or imine cross-links to ensure the nanoparticle maintains its structural integrity. The simultaneous application of both low pH and the reducing agent cleaves both the disulfide and imine cross-links, triggering complete particle disassembly. Nanoparticle disassembly, however, is not necessarily a particularly useful response, and arguably a more desirable response would be the triggered release of a cargo from the nanoparticle. Experiments showed that the application of low pH does cause some cargo escape from these nanoparticles, presumably because the porosity of the particle increases even though the polymer chains remain cross-linked by the disulfide bonds. This work does demonstrate some progress, showing that two orthogonal DCBs can be combined to afford a nanoparticle where two stimuli have to be applied simultaneously to trigger a response, and we anticipate that future systems will possess higher levels of sophistication.
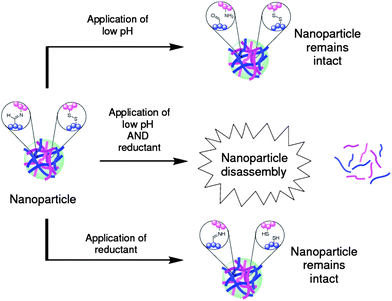 | ||
| Fig. 9 A polymeric nanoparticle requiring the simultaneous application of both lowered pH and a reducing agent to trigger its disassembly into linear polymer chains. Although the application of either lower pH or a reducing agent breaks the imine or disulfide bonds, respectively, there is a sufficiently high density of remaining cross-links to maintain the integrity of the nanoparticle structure.33 | ||
Core cross-linked polymers
CCS polymers are a class of nano-sized polymeric nanoparticle whose structural features are defined by a central core consisting of a network of cross-linked polymer chains surrounded by a corona of polymeric arms.34 Much of the interest in CCS polymers is as a consequence of their architectures, possessing compact structures whose cores can be utilized as carriers for small molecules such as drugs or fragrances, and coronal chains which help to solubilise and shield the cargo from its external environment. CCS polymers also possess solubilities and viscosities similar to linear polymers of relatively low molecular weight, additional properties which make them potentially very useful in a diverse range of fields, most notably drug delivery35 as well as imaging,36 and catalysis.37At Newcastle we became interested in introducing stimuli-responsiveness into CCS polymer architectures using DCBs as cross-links within the cores. Our synthetic strategy borrowed from that described38 by the group of Otsuka and Takahara, which uses the cross-linking of preformed linear diblock copolymer chains. We prepared39 copolymer chains displaying ‘inert’ blocks (Fig. 10) to promote the formation of CCS polymers and discourage macroscopic gel formation, and ‘reactive’ blocks which possess the reaction partners to promote cross-linking through the formation of DCBs. In this work, we focused on using aldehyde and amine functions for eventual cross-linking through imine bond formation, which would allow us to exploit the component exchange capabilities of this bond, and in latter work, its pH-responsiveness. Our initial work demonstrated that imine bond formation was effective in driving the self-assembly of the polymer chains, and we found that the size of the CCS polymers formed could be controlled through the concentration at which the cross-linking was performed, although as the particle size increased so did the particle polydispersity. Particle disassembly could be triggered by addition of a low molecular weight alkyl amine, which undergoes transimination with the imine cross-links within the core. Because this initial work was performed in organic solvents, it was not possible to exploit the pH responsiveness of the imine bond to its full, and so we next turned our attention to water-soluble nanoparticles.
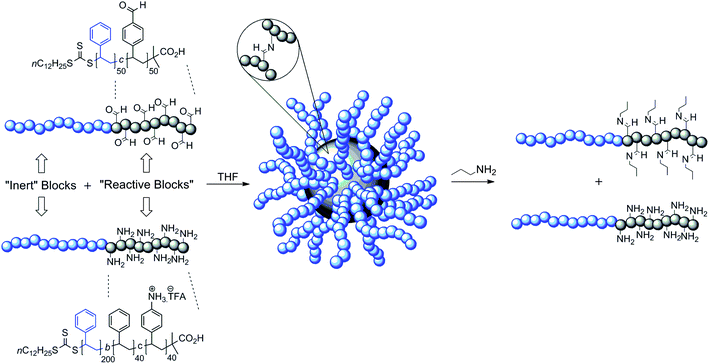 | ||
| Fig. 10 The self-assembly of CCS polymers from diblock copolymers which display either multiple amine or aldehyde functions within one of the blocks. The self-assembly being driven by the formation of imine bond cross-links within the core.39 | ||
This system40 was conceptually similar, but utilized water-soluble acrylamide-based diblock copolymer building blocks to make CCS polymers possessing hydrodynamic radii of 18 nm, and a molecular weigh estimated to be 23 MDa (which corresponds to about 49 arms). These CCS polymers possessed excellent water-solubility, allowing the exploitation of the pH-responsiveness of the imine bonds. Because of the presence of N-isopropylacrylamide monomers within the reactive blocks, the CCS polymers also possessed thermoresponsive cores, and so these CCS polymer species can be considered pH- and temperature-responsive. By changing pH between 11.0 and 5.5, we were able to trigger the assembly/disassembly of the CCS polymer from its component copolymer building blocks, following this process by dynamic light scattering through multiple cycles. By loading the cores of the CCS polymers with a cargo of hydrophobic Nile Red, we were able to show that the pH-induced disassembly process lead to release of the cargo molecules. By holding the pH at 11 and modulating the temperature from 5 °C to 45 °C we were able to show that the release and uptake of the dye molecule could be triggered by changes in temperature. This work hints that conventional stimuli-responsive polymers can be combined with the responsiveness of DCBs to make polymeric species with multiple properties, and although this example can be considered rather simplistic, we are currently exploring more sophisticated dual-responsive systems.
Much of the work discussed so far has focused on exploiting the pH-responsiveness of the imine bond, but of course it should be possible to utilize the other DCBs. Another pH-responsive bond which has very recently been explored within polymeric nanoparticles is the boronic ester, which is formed41 from the condensation of boronic acids with 1,2- or 1,3-diols in aqueous or organic solution (Fig. 4e). Arguably the major application of this reaction has been the use of boronic acids in carbohydrate sensing,42 and a consequence of this research is that much is known about the kinetics and thermodynamics of this reaction. In basic conditions, the reaction between a boronic acid and a hydroxide anion occurs, affording a tetrahedral boron anion which can then reversibly react with a 1,2- or 1,3-diol to form a cyclic boronic ester. In acidic conditions the hydrolysis of the boronic ester back to the boronic acid is favourable. Another interesting feature of boronic acids is that they can trimerize to form a six-membered boroxine in anhydrous organic solvents and in the presence of a Lewis base such as piperidine, a process which is reversible upon the addition of water.
The Jäkle group has prepared43 poly(styreneboronic acid)-b-polystyrene (PSBA-b-PS) diblock copolymers which were shown to non-covalently aggregate in aqueous environments into micellar assemblies on account of the amphiphilicity of the linear diblock copolymers. This micellar system utilises the pH-sensitive sp2 to sp3 interconversion of boronic acids to trigger structural changes in micelle morphology. At high pH, when sp3 hybridization is favourable, the formation of spherical micelles is observed as a consequence of high interface curvature, caused by the electrostatic repulsion of the B(OH)3− groups. At low pH, when sp2 hybridization is favourable, worm-like assemblies were observed as the neutral B(OH)2 functions do not induce any repulsion, reducing the interface curvature. This early boronic acid-containing polymeric system does not utilise pH-sensitive formation of dynamic covalent boronic esters to trigger a structural change, however it does emphasises the versatility and potential of boronic acids in polymeric nanoparticles as a consequence of the pH-induced switch in hybridization.
Building upon their earlier work44 with boronic acid-containing polymers, the Sumerlin research group have focused on utilising the dynamic nature of boronic esters to introduce responsiveness into CCS polymeric nanoparticles. The group has demonstrated45 that boronic acid end-functionalized polymers prepared by RAFT can form three-arm star polymers as a consequence of boroxine cyclisation of the polymer end-groups in the presence of the Lewis base piperidine. The hydrolysis-triggered disassembly back to linear polymer chains was achieved through the addition of water. In subsequent work the Sumerlin group prepared46 (Fig. 11) diblock copolymers containing pendant boronic acid functions within one of the blocks (14), and showed that these linear polymer chains can self-assemble through boronic ester formation with multifunctional small molecule 1,2- or 1,3-diols into CCS polymers (15) possessing hydrodynamic diameters of about 20–30 nm, depending on the actual polymers and cross-linkers used. These CCS polymers possess chemoresponsiveness, as demonstrated by the addition of mono-diol which causes transesterification, triggering disassembly of the star polymer into linear polymer chains. The assembly process can be re-initiated through the addition of further tris-diol, and the Sumerlin group have shown that the assembly/disassembly process can be cycled repeatedly at least six times, a remarkable feat when one considers the build-up of diols in the reaction mixture. Because of the diversity of 1,2- and 1,3-diols in nature, this work opens up the possibility that polymeric nanoparticles whose disassembly can be triggered by the presence of biologically relevant species. One limitation of the current system is its lack of aqueous solubility at physiological pH, although this fact should not present a significant hurdle and which when overcome will also open-up possibilities to exploit the pH-responsive properties of the boronic ester.
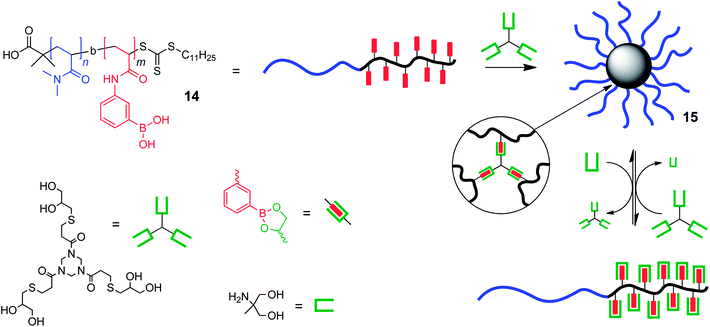 | ||
| Fig. 11 Core cross-linked star polymers are formed via boronic ester formation between pendant boronic acid functions along a diblock copolymer chain and 1,2-diols within multifunctional small molecule cross-linkers.46 | ||
Another class of DCB of interest is the acetal bond,47 which is reversibly formed through the reaction of an aldehyde with two primary alcohols (Fig. 4d). This group is very closely related to the ketal, which is formed through the reaction between a ketone and two primary alcohols, and these functional groups are often prepared in organic solvents under acidic conditions. The resulting acetal/ketal functionality will readily hydrolyse back to its component partners in aqueous media, and the rate of this hydrolysis is pH-sensitive. This pH-responsive feature makes the incorporation of acetal/ketal functional groups within polymeric nanoparticles very appealing, as their acid-catalysed hydrolysis can be utilised to trigger nanoparticle disassembly and subsequent cargo release within low pH environments.48 It is also possible to tune their stabilities by altering the electronic characteristics of the component aldehydes and alcohols. This feature could prove very useful in the field of controlled drug delivery, as certain tissue targets display low pH, such as inflammatory tissues and the phagolysosomes of antigen presenting cells.49 The groups of Haddleton and Davis have developed50 stimuli-responsive biodegradable CCS polymers (Fig. 12) which exploit the pH-responsiveness of ketals. The synthetic approach employed is highly modular, and disulfide cross-linkers can be substituted for the ketal functions allowing CCS polymers which can alternatively exploit the redox-responsiveness of the disulfide linkage. RAFT polymerization techniques were employed to synthesise poly(OEGMA) (16), which forms the coronal arms of the CCS polymers. The polymerization is then continued in the presence of dimethacrylate monomer units to cross-link the growing polymer chains into CCS polymers. Two different cross-linkers have been employed containing either a disulfide (17) or ketal (18) linkage, resulting in the formation two types of CCS polymer possessing either redox- or pH-responsive degradability on account of either disulfide or ketal cross-links present within their cores. These CCS polymers also possess dithiocarbonate functionalities from the retention of the RAFT chain transfer agent, and aminolysis of these dithiocarbonate groups to thiol functions (19) permits post-formation functionalisation through either disulfide bond formation (20) or thiol–ene ‘click’ chemistry51 (21). This feature has been exploited to tag these CCS polymers with fluorescent pyridyl disulfide appendages. The disassembly of the ketal CCS polymers was found to be pH-sensitive, with complete disassembly occurring within 12 h and 1 h, at pH 5.0 and 2.0 respectively. At pH 7.0 the CCS polymers were found to be stable for over 48 h. The disulfide CCS polymers were shown to disassemble over 1 h in the presence of the reducing agent tributyl phosphine. The work demonstrates that well-utilized “arm first” CCS formation techniques can be evolved to introduce dynamic covalent cross-links between the growing polymer chains, and that living radical polymerization techniques introduce the possibility of post-polymerization functionalization at specific sites as a consequence of CTA functionality retention.
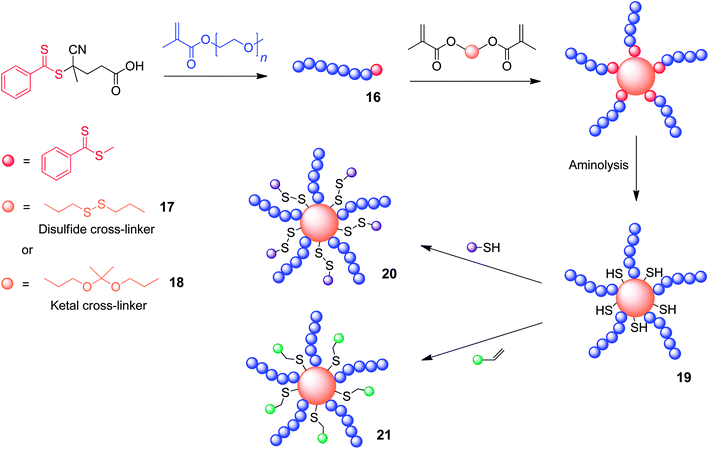 | ||
| Fig. 12 The formation of core cross-linked star polymers through the incorporation of either acetal or disulfide containing dimethacrylate monomer units during polymerization.50 | ||
Covalent bonds which can break at elevated temperatures but reform at ambient temperatures present particularly appealing possibilities when incorporated into polymeric materials. The groups of Otsuka and Takahara have exploited the thermally responsive nature of TEMPO-based alkoxyamine derivatives in the preparation of numerous thermally dynamic polymers and materials, including polymeric nanoparticles. TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) is a stable radical on account of the steric protection afforded by the four adjacent methyl groups, derivatives of which are often referred to as a nitroxyl or nitroxide radicals.52 TEMPO-based alkoxyamine derivatives have been used to mediate polymerizations, furnishing polymers with narrow polydispersity indices and desired end-group functionality, and this particular class of living radical polymerizations (LRP) has been named nitroxide-mediated polymerization (NMP).53
The central C–O bonds within TEMPO-based alkoxyamine derivatives behave as typical covalent bonds under ambient conditions. Upon heating (Fig. 4f) this bond breaks homolytically and a styryl radical and a TEMPO-based radical are formed, and different components can exchange via radical crossover processes. The early work of Otsuka and Takahara in this field focused on preparing54 dynamic covalent polymers chains incorporating thermally dynamic TEMPO-based alkoxyamines within the main chain. These TEMPO-containing monomer units can readily dissociate upon heating and recombine to form polymer chains of varying lengths, demonstrating how linear polymer chains can reconfigure their structural properties at elevated temperatures through thermally exchangeable radical crossover reactions, which then become fixed at ambient conditions. Such thermally-responsive polymers could find application in the field of self-healing materials, and Otsuka and Takahara have recently reviewed the current progress and potential applications in the field of dynamic covalent polymer chains.14c
The group of Otsuka and Takahara have utilised the thermoresponsive nature of TEMPO-based alkoxyamine derivatives to prepare38 core cross-linked star (CCS) polymers (22, Fig. 13). Linear diblock copolymer building blocks (23) which possess an inert block comprised of methyl methacrylate, and a reactive block which contains complimentary pendant alkoxyamine derivatives, have been synthesised by atom-transfer radical polymerizations. These pendant functionalities within the reactive block can undergo radical crossover reactions upon thermal stimulation, which facilitates the cross-linking of multiple polymer chains into CCS polymeric nanoparticles, whilst the inert block discourages macroscopic gelation. These CCS polymers become dynamic when heated and fixed when cooled, and this thermoresponsive nature can be used to trigger disassembly upon the addition of a small molecule TEMPO-based alkoxyamine, which competes through a radical crossover reaction above 110 °C. The groups of Otsuka and Takahara have also exploited this dynamic reaction to cross-link multiple linear polymer chains into star-like nanogel assemblies,55 discrete spherical nanoparticles56 and macroscopically cross-linked organo-57 and hydrogels.58
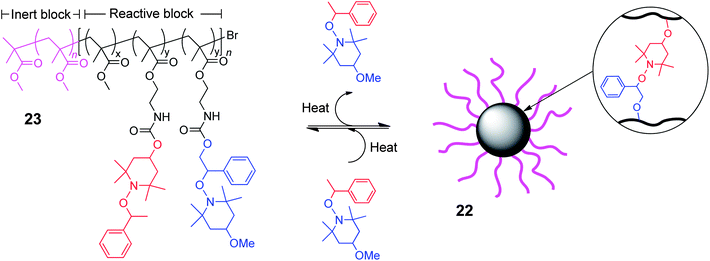 | ||
| Fig. 13 Core cross-linked star polymer formation via a TEMPO-based thermally exchangeable radical crossover reaction between multiple linear copolymer building blocks.38 | ||
Another DCB which is useful in thermally-responsive polymeric materials is the Diels–Alder [4 + 2] cycloaddition reaction between electron-rich dienes and electron-deficient dieneophiles (Fig. 4g). The Diels–Alder reaction of a substituted furan with maleimide forms a Diels–Alder adduct at ∼80 °C, with the reverse reaction being favoured at higher temperatures of ∼150 °C. This reversibility was exploited in spectacular fashion by the Wudl group to prepare59 so-called thermally-mendable polymers, which consist of a macromolecular network formed by cross-linking monomer units through thermally-reversible Diels–Alder reactions. Fractures to this material are likely to involve cleavage of the weakest covalent bonds in the network, namely the Diels–Alder adducts. Fractures can be mended simply by application of a heating/cooling cycle, which causes the Diels–Alder bonds to reform. This landmark work has inspired much effort in the field of mendable polymers utilizing both reversible covalent bonds and non-covalent bonds, and the reader is advised to consult the review60 of Colquhoun and Hayes to learn more. Only recently, however, have researchers begun to look at utilizing thermally reversible Diels–Alder chemistry in the preparation of responsive polymeric nanoparticles. In a particularly elegant study,61 the group of Sumerlin have used Diels–Alder chemistry to cross-link diblock copolymers into thermally responsive core cross-linked star polymers. The diblock copolymers possess appended furan groups within one of the blocks, and cross-linking is performed by heating a solution of polymer at 50 °C with a bis-maleimide cross-linker. Heating a solution of star polymers to 120 °C causes almost complete dissociation of the star polymers back into linear polymer chains, and the group have demonstrated the reversibility of this process by cycling the assembly disassembly process four times. The group of Barthel and Rudolf have also recently exploited62 the thermoreversibility of Diels–Alder adducts to prepare water-soluble core cross-linked micelles which can, at least in part, disassemble back into linear polymer chains at elevated temperatures.
Single-chain polymer nanoparticles
Single-chain polymer nanoparticles are formed from the intramolecular cross-linking of a single polymer chain into a collapsed well-defined nanoparticle whose sizes are typically in the range of 5–20 nm. Such particles are of interest because of their small sizes and their intramolecular cross-links, which are reminiscent of those found in natural biopolymers, in particular proteins. As such they have been proposed by some groups as holding potential as synthetic mimics of the protein folding. Much of the work63 to date in the field of single-chain polymer nanoparticles has involved the application of either kinetically-fixed covalent cross-links, or non-covalent supramolecular interactions such as hydrogen bonding. The dynamic nature of DCBs potentially allows single-chain polymer nanoparticles to possess the chemical robustness of covalent bonds whilst being able to benefit from the dynamic properties of non-covalent bonds. To this end we have developed64 a single chain polymer nanoparticle where a linear polymer chain is intramolecularly cross-linked through dynamic covalent acyl hydrazone bonds. We used a linear styrenic copolymer displaying aromatic aldehyde functions, which was cross-linked with small molecule dihydrazides to form the single chain polymer nanoparticle, and shown how exchange reactions could be used to drive the cross-linking reaction. We believe that such dynamic covalent single-chain polymer nanoparticles which possess adaptable structures may possess the abilities to be ‘molded’ around template molecules, and are further investigating this possibility.The group of Berda have described65 how the redox-responsiveness of the disulfide bond can be used to trigger the ‘folding’ and ‘unfolding’ of polymer chains. This system is based upon (Fig. 14) a linear copolymer (24) prepared by ring-opening olefin metathesis polymerization, which is intramolecularly cross-linked through reaction with the difunctional cross-linker (25) which features a premade disulfide linkage. The ‘unfolding’ of the single-chain polymer nanoparticle can be triggered by reductive cleavage of the disulfide cross-links with dithiothreitol, and this process can be followed by GPC. The ‘folding’ of the linear polymer chain (26) back into the single-chain polymer nanoparticle can be triggered by oxidation with FeCl3. The group hope to expand the scope of this work, and by utilizing multiple orthogonal interactions they aim to be able to program precisely the conformations of single polymer chains.
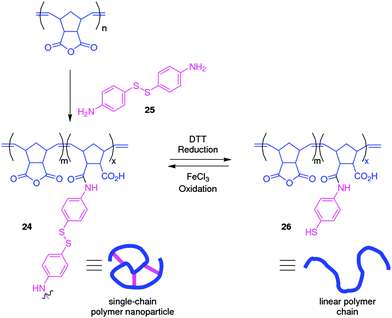 | ||
| Fig. 14 Single chain polymer nanoparticles are formed through the intramolecular cross-linking of polymer chains with a diimine cross-linker. Because of the disulfide bond present within the cross-linker, the nanoparticles are redox responsive and can be reversibly switched to linear polymer chains.65 | ||
Conclusions
The examples covered in this review suggest strongly that DCBs can be used to impart stimuli-responsive features into polymeric nanoparticles, and provide an additional and developing palette of responses which expands that already presented by ‘conventional’ stimuli-responsive polymers. Much of the work to date has focused on the pH-responsiveness of imine or acetal bonds, or the redox responsiveness of the disulfide bond, but research groups are increasingly utilizing other less-explored DCBs such as boronic esters or TEMPO-based alkoxyamines.Much is known about some DCBs with regards to the application of changes in their environments e.g. how the position of the imine equilbria changes with pH, and other less-explored DCBs would benefit from in-depth studies to shed further light on how their equlibria change upon application of a stimulus. The component exchange features of DCBs are also relatively under-exploited within polymeric nanoparticles, and chemists should think more creatively about how this particular feature of DCBs can be exploited. Natural macromolecules are constantly reshuffling and changing their components, resulting in a change of function. For instance, proteins can change their function depending on their glycosylation patterns.66 DCBs can be utilized to mimic such features, allowing polymer nanoparticles to also change their components and thus their function, but we need to be more imaginative in our exploitation of this possibility.
Our own beliefs are that the future direction of this field will be along multiple, but certainly not mutually exclusive directions. We believe that future work has to become more applications driven, taking the proof-of-principle knowledge gained so-far and exploiting it to develop stimuli-responsive nanoparticles tailored for particular applications. For example, many responsive polymeric nanoparticles are described as having “potential” in drug delivery, but to make a real impact in this field one must tailor a system to deliver a specific drug to a specific tissue/cell type, working in collaboration with pharmacologists and other experts to overcome the many significant challenges which must be addressed to develop systems possessing high in vivo efficacy. Researchers should also consider more seriously the development of polymer nanoparticles which respond to other less explored stimuli such as pressure, electrical or near-infrared, which would also drive the field onwards and open-up new possibilities in terms of possible applications. DCBs may only play a part in more sophisticated nanoparticles, and it is likely that DCBs will enhance or work in synergy with the ‘conventional’ stimuli-responsive polymers to provide nanoparticles whose responsive natures are more than the sum of their parts. The application of combinations of orthogonal DCBs is another way in which the levels of sophistication will likely be increased.
Many of the polymeric nanoparticles described here are constructed from aggregates of polymer chains. This approach allows the exploitation of the spectacular advances in polymerization methodologies, in particular living radical polymerizations, to prepare high-precision polymer building blocks for the construction of polymer nanoparticles. It must be remembered, however, that emulsion polymerization methods are still by far the industrially most important way to make polymeric nanoparticles, and chemists should think further on how DCBs can be incorporated into these important classes of nanoparticles. In this regard, a promising example reported67 by the groups of Colson and Grinstaff are the so-called “expansive” nanoparticles, which are prepared using miniemulsion techniques. These nanoparticles contain pH-sensitive acetal linkages, which when cleaved cause the nanoparticle to switch from hydrophobic to a hydrophilic hydrogel. As water enters this nanoparticle, it undergoes a several hundred-fold increase in size and releases encapsulated drug. This class of nanoparticle possesses considerable potential in drug delivery, and Colson and Grinstaff have written an excellent review article68 which the reader is advised to consult to learn more about this unique class of responsive nanoparticles.
With sufficient imagination, creativity and tenacity, it is the opinion of these authors that new responsive polymeric nanoparticles will be developed which will go on to make big impacts on the world around us. It is our hope that this review may convince readers that this field is an area of much promise.
References
- (a) Colloids in Paints, ed. T. F. Tadros, Wiley-VCH Verlag GmbH & Co, KGaA, Weinheim, 2010 Search PubMed; (b) Paints, Coatings and Solvents, ed. W. Freitag and D. Stoy, Wiley-VCH Verlag GmbH & Co, KGaA, Weinheim, 2nd edn, 1998 Search PubMed; (c) J. W. Taylor and M. A. Winnik, J. Coat. Technol. Res., 2004, 1, 163–190 CrossRef CAS.
- (a) M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 7710782 CrossRef; (b) J. V. Jokerst and S. S. Gambir, Acc. Chem. Res., 2011, 44, 1050–1060 CrossRef CAS; (c) N. Kamaly, Z. Xiao, P. M. Valencia, A. F. Radovic-Moreno and O. C. Farokhzad, Chem. Soc. Rev., 2012, 41, 2971–3010 RSC.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS.
- C. D. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- M. D. C. Topp, P. J. Dijkstra, H. Talsma and J. Feijen, Macromolecules, 1997, 30, 8518–8520 CrossRef CAS.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- (a) C. M. Schilli, M. F. Zhang, E. Rizzardo, S. H. Thang, Y. K. Chong, K. Edwards, G. Karlsson and A. H. E. Muller, Macromolecules, 2004, 37, 7861–7866 CrossRef CAS; (b) F. Kohori, K. Sakai, T. Aoyagi, M. Yokoyama, Y. Sakurai and T. Okano, J. Controlled Release, 1998, 55, 87–98 CrossRef CAS.
- (a) E. Chung, M. Yokoyama, M. Yamato, T. Aoyagi, Y. Sakurai and T. Okano, J. Controlled Release, 1999, 62, 115–127 CrossRef; (b) J. C. Leroux, E. Roux, D. Le Garrec, K. L. Hong and D. C. Drummond, J. Controlled Release, 2001, 72, 71–84 CrossRef CAS; (c) S. H. Qin, Y. Geng, D. E. Discher and S. Yang, Adv. Mater., 2006, 18, 2905–2909 CrossRef CAS.
- (a) L. A. Lyon, Z. Y. Meng, N. Singh, C. D. Sorrell and A. S. John, Chem. Soc. Rev., 2009, 38, 865–874 RSC; (b) M. Nakayama and T. Okano, J. Drug Delivery Sci. Technol., 2006, 16, 35–44 CAS; (c) H. Wei, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, Prog. Polym. Sci., 2009, 34, 893–910 CrossRef CAS.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed., 1996, 35, 1155–1196 CrossRef CAS.
- (a) J.-M. Lehn, Aust. J. Chem., 2010, 63, 611–623 CrossRef CAS; (b) J.-M. Lehn, Chem. Soc. Rev., 2007, 37, 151–160 RSC; (c) J.-M. Lehn, Prog. Polym. Sci., 2005, 30, 814–831 CrossRef CAS.
- (a) R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS; (b) C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS; (c) T. Maeda, H. Otsuka and A. Takahara, Prog. Polym. Sci., 2009, 34, 581–604 CrossRef CAS.
- (a) The Chemistry of the Carbon–Nitrogen Double Bond, ed. S. Patai, Interscience, London, 1968 Search PubMed; (b) C. D. Meyer, C. S. Joiner and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 1705–1723 RSC; (c) M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- C. Godoy-Alcántar, A. K. Yatsimirsky and J.-M. Lehn, J. Phys. Org. Chem., 2005, 18, 979–985 CrossRef.
- (a) W. P. Jencks, J. Am. Chem. Soc., 1959, 81, 475–481 CrossRef CAS; (b) W. P. Jencks, Catalysis in Chemistry and Enzymology, Dover Publications, Inc., New York, 1986, pp. 467–471 Search PubMed.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J. L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS.
- (a) C. Booth, T. D. Naylor, C. Price, N. S. Rajab and R. B. Stubbersfield, J. Chem. Soc., Faraday Trans. 1, 1978, 74, 2352–2362 RSC; (b) C. Price, Pure Appl. Chem., 1983, 55, 1563–1572 CrossRef CAS; (c) P. Bahadur, S. V. Sastry, S. Marti and G. Reiss, Colloids Surf., 1985, 16, 337–346 CrossRef CAS.
- (a) A. Guo, G. J. Liu and J. Tao, Macromolecules, 1996, 29, 2487–2493 CrossRef CAS; (b) J. Tao, S. Stewart, G. J. Liu and M. L. Yang, Macromolecules, 1997, 30, 2738–2745 CrossRef CAS; (c) F. Henselwood and G. J. Liu, Macromolecules, 1998, 31, 4213–4217 CrossRef CAS; (d) Y. Y. Won, H. T. Davis and F. S. Bates, Science, 1999, 283, 960–963 CrossRef CAS.
- E. S. Read and S. Armes, Chem. Commun., 2007, 3021–3035 RSC.
- X. W. Xu, J. D. Flores and C. L. McCormick, Macromolecules, 2011, 44, 1327–1334 CrossRef CAS.
- C. B. Minkenberg, L. Florusse, R. Eelkema, G. J. M. Koper and J. H. J. van Esch, J. Am. Chem. Soc., 2009, 131, 11274–11275 CrossRef CAS.
- C. Wang, G. Wang, Z. Wang and X. Zhang, Langmuir, 2011, 27, 12375–12380 CrossRef.
- A. W. Jackson and D. A. Fulton, Macromolecules, 2010, 43, 1069–1075 CrossRef CAS.
- L. He, Y. Jiang, C. Tu, G. Li, B. Zhu, C. Jin, Q. Zhu, D. Yan and X. Zhu, Chem. Commun., 2010, 46, 7569–7571 RSC.
- C. Wang, G. Wang, Z. Wang and X. Zhang, Chem.–Eur. J., 2011, 17, 3322–3325 CrossRef CAS.
- (a) R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS; (b) M. L. Adams, A. Layasanifar and G. S. Kwon, J. Pharm. Sci., 2003, 92, 1343–1355 CrossRef CAS.
- (a) J. H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227–17235 CAS; (b) S. Jiwpanich, J. H. Ryu, S. Bickerton and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 10683–10685 CrossRef CAS; (c) J. H. Ryu, S. Jiwpanich, R. Chacko, S. Bickerton and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 8246–8247 CrossRef CAS.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS.
- A. Meister and M. E. Anderson, Annu. Rev. Biochem., 1983, 52, 711–760 CrossRef CAS.
- J. Dai, S. Lin, D. Cheng, S. Zou and X. Shuai, Angew. Chem., Int. Ed., 2011, 50, 9404–9408 CrossRef CAS.
- A. W. Jackson and D. A. Fulton, Macromolecules, 2012, 45, 2699–2708 CrossRef CAS.
- H. Gao, Macromol. Rapid Commun., 2012, 33, 722–734 CrossRef CAS; H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef; A. Blencowe, J. F. Tan, T. K. Goh and G. G. Qiao, Polymer, 2009, 50, 5–32 CrossRef.
- J. T. Wiltshire and G. G. Qiao, Aust. J. Chem., 2007, 60, 699–705 CrossRef CAS.
- K. Fukukawa, R. Rossin, A. Hagooly, E. Pressly, J. Hunt, B. Messmore, K. Wooley, M. Welch and C. J. Hawker, Biomacromolecules, 2008, 9, 1329–1339 CrossRef CAS.
- (a) T. Terashima, M. Kamigaito, K.-Y. Baek, T. Ando and M. Sawamoto, J. Am. Chem. Soc., 2003, 125, 5288–5289 CrossRef CAS; (b) B. Helms, S. J. Guillaudeu, Y. Xie, M. McMurdo, C. J. Hawker and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2005, 44, 6384–6387 CrossRef CAS; (c) T. Terashima, M. M. Ouchi, T. Ando, M. Kamigaito and M. Sawamoto, Macromolecules, 2007, 40, 3581–3588 CrossRef CAS.
- Y. Amamoto, Y. Higaki, Y. Matsuda, H. Otsuka and A. Takahara, J. Am. Chem. Soc., 2007, 129, 13298–13304 CrossRef CAS.
- A. W. Jackson and D. A. Fulton, Chem. Commun., 2010, 46, 6051–6053 RSC.
- A. W. Jackson and D. A. Fulton, Chem. Commun., 2011, 47, 6807–6809 RSC.
- T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Angew. Chem., Int. Ed. Engl., 1996, 35, 1911–1922 CrossRef CAS.
- W. Wang, X. M. Gao and B. H. Wang, Curr. Org. Chem., 2002, 6, 1285–1317 CrossRef CAS; T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Nature, 1995, 374, 345–347 CrossRef.
- Y. Qin, V. Sukul, D. Pagakos, C. Z. Cui and F. Jäkle, Macromolecules, 2005, 38, 8987–8990 CrossRef CAS.
- (a) J. N. Cambre, D. Roy, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2007, 129, 10348–10349 CrossRef CAS; (b) D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2008, 2477–2479 RSC; (c) D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2009, 2106–2108 RSC.
- P. De, S. R. Gondi, D. Roy and B. S. Sumerlin, Macromolecules, 2009, 42, 5614–5621 CrossRef CAS.
- A. P. Bapat, D. Roy, J. G. Ray, D. A. Savin and B. S. Sumerlin, J. Am. Chem. Soc., 2011, 133, 19832–19838 CrossRef CAS.
- J. F. Stoddart, Stereochemistry of Carbohydrate Compounds, Wiley Interscience, New York, 1971 Search PubMed.
- For some recent relevant examples of polymeric nanoparticles containing acid-sensitive acetal/ketal linkages see: (a) S. J. Lee, K. H. Min, H. J. Lee, A. N. Koo, H. P. Rim, B. J. Jeon, S. Y. Jeong, J. S. Heo and S. C. Lee, Biomacromolecules, 2011, 12, 1224–1233 CrossRef CAS; (b) N. Murthy, Y. X. Thng, S. Schuck, M. C. Xu and J. M. J. Frechet, J. Am. Chem. Soc., 2002, 124, 12398–12399 CrossRef CAS; (c) Y. Chan, T. Wong, F. Byrne, M. Kavallaris and V. Bulmus, Biomacromolecules, 2008, 9, 1826–1836 CrossRef CAS; (d) Y. L. Li, W. J. Du, G. R. Sun and K. L. Wooley, Macromolecules, 2008, 41, 6605–6607 CrossRef CAS.
- (a) G. Helmlinger, A. Schell, M. Dellian, N. S. Forbes and R. K. Jain, Clin. Cancer Res., 2002, 8, 1284–1291 CAS; (b) A. S. Trevani, G. Andonegui, M. Giordano, D. H. Lopez, R. Gamberale, F. Minucci and J. R. Geffner, J. Immunol., 1999, 162, 4849–4857 CAS.
- J. A. Syrett, D. M. Haddleton, M. R. Whittaker, T. P. Davis and C. Boyer, Chem. Commun., 2011, 47, 1449–1451 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- (a) A. L. J. Beckwith, V. W. Bowry and K. U. Ingold, J. Am. Chem. Soc., 1992, 114, 4983–4992 CrossRef CAS; (b) V. W. Bowry and K. U. Ingold, J. Am. Chem. Soc., 1992, 114, 4992–4996 CrossRef CAS; (c) I. W. C. E. Arends, P. Mulder, K. B. Clark and D. D. M. Wayner, J. Phys. Chem., 1995, 99, 8182–8189 CrossRef CAS.
- (a) C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS; (b) M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS; (c) D. Greszta and K. Matyjaszewski, Macromolecules, 1996, 29, 7661–7670 CrossRef CAS; (d) C. J. Hawker, G. G. Barclay, A. Orellana, J. Dao and W. Devonport, Macromolecules, 1996, 29, 5245–5254 CrossRef CAS.
- H. Otsuka, K. Aotani, Y. Higaki and A. Takahara, Chem. Commun., 2002, 2838–2839 RSC.
- Y. Amamoto, T. Maeda, M. Kikuchi, H. Otsuka and A. Takahara, Chem. Commun., 2009, 689–691 RSC; Y. Amamoto, M. Kikuchi, H. Otsuka and A. Takahara, Polym. J., 2010, 42, 860–867 CrossRef CAS; Y. Amamoto, M. Kikuchi, H. Otsuka and A. Takahara, Macromolecules, 2010, 43, 5470–5473 CrossRef; Y. Amamoto, M. Kikuchi, H. Masunaga, S. Sasaki, H. Otsuka and A. Takahara, Macromolecules, 2010, 43, 1785–1791 CrossRef.
- Y. Higaki, H. Otsuka and A. Takahara, Macromolecules, 2006, 39, 2121–2125 CrossRef CAS.
- Y. Amamoto, M. Kikuchi, H. Masunaga, S. Sasaki, H. Otsuka and A. Takahara, Macromolecules, 2009, 42, 8733–8738 CrossRef CAS.
- J. Su, Y. Amamoto, M. Nishihara, A. Takahara and H. Otsuka, Polym. Chem., 2011, 2, 2021–2026 RSC.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS.
- S. Burattini, B. W. Greenland, D. Chappell, H. M. Colquhoun and W. Hayes, Chem. Soc. Rev., 2010, 39, 1973–1985 RSC.
- A. P. Bapat, J. G. Ray, D. A. Savin, E. A. Hoff, D. L. Patton and B. S. Sumerlin, Polym. Chem., 2012, 3, 3112–3120 RSC.
- M. J. Barthel, T. Rudolph, S. Crotty, F. H. Schacher and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2012 DOI:10.1002/pola.26327.
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2012, 33, 958–971 CrossRef CAS; M. Aiertza, I. Odriozola, G. Cabañero, H.-J. Grande and I. Loinaz, Cell. Mol. Life Sci., 2012, 69, 337–346 CrossRef.
- B. S. Murray and D. A. Fulton, Macromolecules, 2011, 44, 7242–7252 CrossRef CAS.
- B. T. Tuten, D. Chao, C. K. Lyon and E. B. Berda, Polym. Chem., 2012, 3, 3068–3071 RSC.
- Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Cold Spring Harbor laboratory Press, Cold Spring Harbor, New York, 2nd edn, 2009 Search PubMed.
- A. P. Griset, J. Walpole, R. Liu, A. Gaffey, Y. L. Colson and M. W. Grinstaff, J. Am. Chem. Soc., 2009, 131, 2469–2471 CrossRef CAS.
- Y. L. Colson and M. W. Grinstaff, Adv. Mater., 2012, 28, 3878–3886 CrossRef.
| This journal is © The Royal Society of Chemistry 2013 |