Facilely prepared inexpensive and biocompatible self-healing hydrogel: a new injectable cell therapy carrier†
Bin
Yang
a,
Yaling
Zhang
a,
Xiaoyong
Zhang
a,
Lei
Tao
*a,
Shuxi
Li
a and
Yen
Wei
*ab
aDepartment of Chemistry, Tsinghua University, Beijing 100084, P. R. China. E-mail: leitao@mail.tsinghua.edu.cn; Tel: +86-010-62792604
bKey Lab of Organic Optoelectronic & Molecular Engineering of Ministry of Education, Department of Chemistry, Tsinghua University, Beijing 100084, P. R. China. E-mail: weiyen@tsinghua.edu.cn
First published on 24th August 2012
Abstract
An inexpensive, biocompatible self-healing hydrogel as a new injectable cell therapy carrier has been facilely developed.
Injectable hydrogels could accommodate irregular shaped defects and remain at the desired position by implanting into tissues through a minimally invasive strategy.1–3 Some stimuli-responsive injectable hydrogels have been successfully developed. Typically, the hydrogel precursors are injected as liquid and the sol–gel processes are triggered in situ by changing physical/chemical conditions, such as by varying temperature, pH, ionic strength or by redox-initiated, photo-initiated polymerizations, chemical/enzyme catalytic cross-linking reactions and so on.4–10 The in situ formed hydrogel could remain at a desired position for controlled-release of encapsulated therapeutics.11 Thus, injectable hydrogels have attracted much research attention for their potential applications for cell therapy and drug delivery.12,13 However, these traditional injectable hydrogels are generated through drastic change of environmental conditions or using some toxic organic reagents, which will more or less detrimentally affect the encapsulated drugs or cells.14 Meanwhile, the slow gelation might result in the cargo loss and diffusion from the targeting site while the extremely rapid gelation process might lead to the undesired premature polymerization and delivery failure.15,16
Synthetic materials that are capable of autonomous healing upon damage have been developed rapidly because of their many potential applications.17 As a new type of biomaterial, self-healing hydrogels have been considered as an alternative to traditional injectable hydrogels.18,19 Compared with traditional injectable hydrogels, self-healing hydrogels could homogeneously encapsulate pharmaceutical drugs/cells ex vivo under physiology conditions, resulting in better repeatable experimental results and higher therapeutic activities of drugs and cells.20 After injection, the broken hydrogel fragments could regenerate an integral gel at the target site, avoiding the risk of catheter clogging by premature polymerization.
Although many new types of self-healing hydrogels have been successfully developed, the major self-healing hydrogels for biomedical applications are based on the self-assembly of amphiphilic peptides,18,21 which are normally expensive and complicated to prepare since those peptides need to be artificially designed and synthesized using an automated solid phase peptide synthesizer,22 hindering the large-scale manufacture and counteracting the advantages of those self-healing hydrogels. Thus, preparing a biocompatible self-healing hydrogel using inexpensive gelators through a facile method is critical for the practical application of self-healing hydrogels.
Recently, we developed a facile approach to prepare a self-healing hydrogel using cheap chitosan and easily synthesized biocompatible telechelic difunctional poly(ethylene glycol) (DF-PEG) as main components.23,24 DF-PEG was prepared by esterification of hydroxyl terminated PEG with 4-formylbenzoic acid. By just simply mixing chitosan and DF-PEG solutions, a hydrogel could be prepared at room temperature in less than 1 min. The hydrogel 3D network is constructed through dynamic covalent Schiff-base linkage between NH2 groups on chitosan and benzaldehyde groups at PEG chain ends. Due to the intrinsic dynamic equilibrium, the Schiff-base could be considered as a quasi-covalent linkage,25–27 the cleavage and regeneration of the imine bond keep occurring in the hydrogel network. As a result, the hydrogel could self-heal itself automatically without additional stimuli. Additionally, many bio-stimuli, such as enzymes, amino acids, vitamin B6 derivatives and pH could shift the dynamic equilibrium to disaggregate the hydrogel, suggesting the potential application of this hydrogel for controlled-release of therapeutics.
In the current work, we hope to evaluate the possibility of using this inexpensive, facilely prepared, biocompatible hydrogel as a new injectable cell therapy carrier. The synthetic strategy we applied is summarized in Scheme 1. Glycol chitosan (GCS) instead of chitosan was used for its better solubility under physiological conditions. After mixing GCS and DF-PEG4000 solutions, a hydrogel has been facilely prepared at 25 °C within 1 min.
 | ||
| Scheme 1 Preparation of cells encapsulated in a self-healing hydrogel under physiology conditions. | ||
The GCS-based hydrogel could form quickly through dynamic Schiff-base crosslinkage between amine groups of GCS and benzaldehyde groups on DF-PEG termini (Fig. 1A, ∼150 s). Meanwhile, the storage modulus G′ and loss modulus G′′ can be adjusted by changing the wt% ratio of gelators. A series of hydrogels with the same wt% GCS (1.5 wt%) and different wt% DF-PEG4000 (0.56 wt%, 1.1 wt%, 2.2 wt%) were prepared. As expected, the higher storage modulus G′ value was observed with the higher wt% DF-PEG4000 (Fig. 1B).
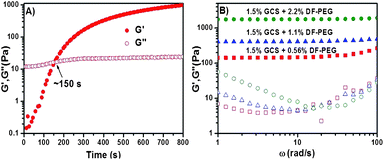 | ||
| Fig. 1 Rheology analyses of the (A) gelation process; (B) storage modulus G′ (solid) and loss modulus G′′ (hollow) of the hydrogels with the same wt% glycol chitosan and different wt% DF-PEG4000 (green: 2.2 wt%; blue: 1.1 wt%; red: 0.56 wt%). Sweeps were performed at 1% strain and 6.3 rad s−1. | ||
Since the hydrogel network is constructed through Schiff-base, transient linkages between GCS and DF-PEG4000, the polymer chains in the hydrogel network are expected to de-crosslink and re-crosslink ceaselessly, resulting in self-healability of the hydrogel. Thus, after injection, the hydrogel should deform under shear stress and recover back into a whole hydrogel when stress is removed (self-healable). To test that, a dynamic GCS–PEG hydrogel (stained with trypan blue for easy observation) was prepared in a syringe barrel. As a control, a gelatin hydrogel (stained with rhodamine B) was prepared in another syringe barrel (Fig. 2A-1). These two hydrogels were extruded directly through 21-gauge needles into water-containing bottles, respectively (Fig. 2A-2). The difference between the self-healing hydrogel and the conventional hydrogel could be clearly observed. After injection, the broken GCS–PEG hydrogel pieces regenerated a completely homogeneous hydrogel in 30 minutes (Fig. 2A-3, right and A-4). In sharp contrast, the gelatin hydrogel still remained as fragments (Fig. 2A-3, left), demonstrating the excellent self-healability of the GCS–PEG hydrogel. An additional self-healing experiment was carried out by punching a hole on both the self-healable and the gelatin hydrogel. The artificially punched hole in the control gel (conventional 5% gelatin hydrogel) remained for the time period of 15 min. In sharp contrast, the hole in the self-healable gel closed up gradually and finally disappeared within ∼15 min (Fig. S1†).
 | ||
| Fig. 2 (A) Self-healing process after injection (blue: self-healing hydrogel, red: gelatin hydrogel). (B) Rheology analyses of hydrogel deformation and recovery. Storage modulus G′ (filled symbols) and loss modulus G′′ (empty symbols) of 1.5 wt% glycol chitosan + 2.2 wt% DF-PEG4000 gels evolving over time from repeated cycles of 3 min low 1% strain and 2 min high 200% strain oscillations at 6.3 rad s−1. (C) G′ and G′′ versus time during the self-healing process (frequency: 1.0 Hz; strain: 1.0%). (D) Storage modulus G′ and loss modulus G′′ of original (1.5 wt% GCS + 2.2 wt% DF-PEG) and self-healed hydrogels. | ||
Rheology analyses were further carried out to quantitatively monitor the self-healing process. In brief, the profile of G′ and G′′ values to different amplitudes was shown in Fig. 2B. Amplitude oscillatory forces were changed from γ = 200% to 1% under the same frequency (1.0 Hz) to test the recovery of mechanical properties of the hydrogel, and the process was repeated twice. When the hydrogel was analysed with a large amplitude oscillatory force (Fig. 2B, γ = 200%, frequency = 1.0 Hz), the G′ value decreased from ∼1000 to ∼200 Pa, indicating a loose hydrogel network (tan δ ≡ G′′/G′ ≈ 0.7 to 1.0). After decreasing the amplitude (γ = 1%, frequency = 1.0 Hz), the G′ recovered quickly to the initial value, and the hydrogel returned to the original state (tan δ ≈ 0.01), indicating the quick recovery of the hydrogel inner network and confirming the self-healing capability of the GCS–PEG hydrogel.
Additionally, another rheology analysis was carried out to monitor qualitatively the self-healing process. In brief, a gel was prepared as described above (1.5 wt% GCS and 2.2 wt% DF-PEG) and tested for the storage modulus G′ (∼2000 Pa, Fig. 2D). The gel was subsequently cut into 9 pieces on a plate, and the broken hydrogel exhibited lower storage modulus G′ (∼390 Pa, Fig. 2C) than the original one. However, the G′ value increased with time and finally reached a similar value to that of the original hydrogel. The G′ and G′′ values of the self-healed gel versus frequency were almost the same as those of the original hydrogel (Fig. 2D), indicating qualitatively the recovery of the inner structure of the hydrogel.
Biocompatibility is critical for biomaterials' application. Thus, HeLa cells have been used to test the cytotoxicity of the synthesized DF-PEG4000. As shown in Fig. S2,† no obvious cell viability decrease was observed via cell viability examination. Even when the concentration of DF-PEG4000 reached up to 9.0 mg mL−1, the cell viability was still greater than 80%. Compared with PEG4000, the introduction of a benzaldehyde group seems not to bring about obvious cytotoxicity to the polymer. Given GCS is a well-known biocompatible biopolymer,28–30 all the components in the hydrogel are almost non-toxic, suggesting the excellent biocompatibility of our GCS–PEG hydrogel.
As a cell therapy carrier, cells should live well in the hydrogel. To test that, HeLa cells were suspended in RPMI-1640 media and mixed with GCS dissolved in the same media. The HeLa–GCS suspension was pipetted into a Petri-dish, and then DF-PEG4000 in RPMI-1640 media was pipetted into the same dish and gently mixed to induce gel formation. As shown in Fig. 3, fluorescent confocal microscopy images of cells stained with the fluorescein diacetate/propidium iodide (FDA/PI) reagent illustrate that HeLa cells tolerated the 3D encapsulation in the hydrogels (∼97% viability after 24 h) and lived well in the hydrogel network (∼87% viability after 72 h). Such a long survival period in the hydrogel is crucial for the injected cells to play their therapeutic role at the targeting position.
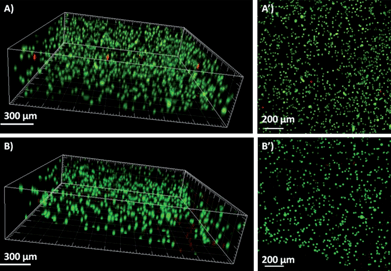 | ||
| Fig. 3 3D (A and B) and z-axis maximum projection (A′ and B′) views of confocal microscopy images. Cell viability (viable cells: green, dead cells: red) and spatial distribution of HeLa cells encapsulated in the self-healing hydrogels. (A) 24 h culture after gelation. (B) 72 h culture after gelation. | ||
The primary injection experiment of cell encapsulated hydrogel was subsequently carried out. The cell containing hydrogel was prepared in a syringe and then pushed through a 21-gauge needle into a Petri-dish. As shown in Fig. 4, fluorescent confocal microscopy images of cells stained with the FDA/PI reagent illustrated that HeLa cells could tolerate 3D encapsulation, the subsequent injection and self-healing processes. There are ∼87% live cells after the injection and self-healing processes (Fig. 4A, 2 h). After 24 h, ∼85% of the cells still retained high viability in the hydrogel. Some more dead cells were observed after injection, however, the dead cell amount did not increase during a 24 h culture, indicating that most dead cells might be a result of the extrusion process in the needle.
 | ||
| Fig. 4 Cell delivery in the hydrogels. 3D (A and B) and z-axis maximum projection (A′ and B′) views of confocal microscopy images. The viability (viable cells: green, dead cells: red) and spatial distribution of HeLa cells (A) 2 h and (B) 24 h after encapsulated in the gels after injecting into a Petri-dish. | ||
Conclusions
A glycol chitosan-based hydrogel has been facilely prepared by mixing GCS and DF-PEG4000 solutions under benign conditions. After injected through a 21-gauge needle, the broken hydrogel pieces could recover back into a complete homogeneous hydrogel. The cytotoxicity evaluation of the DF-PEG indicated that the telechelic PEG is almost nontoxic. Additionally, cells could tolerate the 3D hydrogel encapsulation and subsequent injection and self-healing processes, suggesting this GCS–PEG hydrogel might have potential for the 3D cell culture and injection cell therapy. Considered the inexpensive biocompatible gelators and the facile approach to prepare the hydrogel, this self-healing hydrogel could greatly reduce the cost and increase the feasibility of cell therapy. The in vivo evaluation and application of this novel self-healing hydrogel is carried out in our continued research.Acknowledgements
This research was supported by the National Science Foundation of China (21104039, 21134004) and the National 973 Project (no. 2011CB935700).Notes and references
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473 RSC.
- H. Tan and K. G. Marra, Materials, 2010, 3, 1746 CrossRef CAS.
- M. K. Nguyen and D. S. Lee, Macromol. Biosci., 2010, 10, 563 CrossRef CAS.
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860 CrossRef CAS.
- J. F. Mano, Adv. Eng. Mater., 2008, 10, 515 CrossRef CAS.
- W. A. Petka, J. L. Harden, K. P. McGrath, D. Wirtz and D. A. Tirrell, Science, 1998, 281, 389 CrossRef CAS.
- H. J. Chung and T. G. Park, Nano Today, 2009, 4, 429 CrossRef CAS.
- L. Zhao, L. Zhu, F. Liu, C. Liu, Q. Wang, C. Zhang, J. Li, J. Liu, X. Qu and Z. Yang, Int. J. Pharm., 2011, 410, 83 CrossRef CAS.
- W. S. Shim, J. S. Yoo, Y. H. Bae and D. S. Lee, Biomacromolecules, 2005, 6, 2930 CrossRef CAS.
- Q. Li, J. Wang, S. Shahani, D. D. N. Sun, B. Sharma, J. H. Elisseeff and K. W. Leong, Biomaterials, 2006, 27, 1027 CrossRef CAS.
- B. D. Ratner and S. J. Bryant, Annu. Rev. Biomed. Eng., 2004, 6, 41 CrossRef CAS.
- M. K. Nguyen and D. S. Lee, Chem. Commun., 2010, 46, 3583 RSC.
- M. Kurisawa, J. E. Chung, Y. Y. Yang, S. J. Gao and H. Uyama, Chem. Commun., 2005, 4312 RSC.
- G. Molinaro, J. C. Leroux, J. Damas and A. Adam, Biomaterials, 2002, 23, 2717 CrossRef CAS.
- D. Gupta, C. H. Tator and M. S. Shoichet, Biomaterials, 2006, 27, 2370 CrossRef CAS.
- T. P. Martens, A. F. G. Godier, J. J. Parks, L. Q. Wan, M. S. Koeckert, G. M. Eng, B. I. Hudson, W. Sherman and G. Vunjak-Novakovic, Cell Transplant., 2009, 18, 297 CrossRef.
- A. Phadke, C. Zhang, B. Arman, C. C. Hsu, R. A. Mashelkar, A. K. Lele, M. J. Tauber, G. Arya and S. Varghese, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4383 CrossRef CAS.
- C. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528 RSC.
- M. Guvendiren, H. D. Lu and J. A. Burdick, Soft Matter, 2012, 8, 260 RSC.
- C. Yan, A. Altunbas, T. Yucel, R. P. Nagarkar, J. P. Schneider and D. J. Pochan, Soft Matter, 2010, 6, 5143 RSC.
- I. Hamley, Soft Matter, 2011, 7, 4122 RSC.
- H. D. Lu, M. B. Charati, I. L. Kim and J. A. Burdick, Biomaterials, 2012, 33, 2145 CrossRef CAS.
- Y. Zhang, L. Tao, S. Li and Y. Wei, Biomacromolecules, 2011, 12, 2894 CrossRef CAS.
- Y. L. Zhang, B. Yang, X. Y. Zhang, L. X. Xu, L. Tao, S. X. Li and Y. Wei, Chem. Commun., 2012, 48, 9305–9307 RSC.
- M. W. Urban, Nat. Chem., 2012, 4, 80 CrossRef CAS.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898 CrossRef.
- T. Maeda, H. Otsuka and A. Takahara, Prog. Polym. Sci., 2009, 34, 581 CrossRef CAS.
- J. H. Park, Y. W. Cho, H. Chung, I. C. Kwon and S. Y. Jeong, Biomacromolecules, 2003, 4, 1087 CrossRef CAS.
- J. Hyung Park, S. Kwon, M. Lee, H. Chung, J. H. Kim, Y. S. Kim, R. W. Park, I. S. Kim, S. Bong Seo and I. C. Kwon, Biomaterials, 2006, 27, 119 CrossRef.
- H. Sashiwa, N. Kawasaki, A. Nakayama, E. Muraki, N. Yamamoto and S. Aiba, Biomacromolecules, 2002, 3, 1126 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental description and results including cytotoxicity evaluation, etc. See DOI: 10.1039/c2py20627g |
| This journal is © The Royal Society of Chemistry 2012 |