Effect of TiO2 nanoparticle surface functionalization on protein adsorption, cellular uptake and cytotoxicity: the attachment of PEG comb polymers using catalytic chain transfer and thiol–ene chemistry†
Roslyn
Tedja
a,
Alexander H.
Soeriyadi
b,
Michael R.
Whittaker
b,
May
Lim
a,
Christopher
Marquis
*c,
Cyrille
Boyer
*b,
Thomas P.
Davis
b and
Rose
Amal
a
aARC Centre of Excellence for Functional Nanomaterials, School of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia
bCentre for Advanced Macromolecular Design, School of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: cboyer@unsw.edu.au; Fax: +61 2 9385 5966; Tel: +61 2 9385 7955
cSchool of Biotechnology and Biomolecular Sciences, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: c.marquis@unsw.edu.au; Fax: +61 2 9385 1483; Tel: +61 29385 3898
First published on 31st July 2012
Abstract
A successful modification of titanium dioxide (TiO2) nanoparticles surfaces by a grafting-to polymer technique combining catalytic chain transfer and thiol–ene click chemistry is reported. Vinylic end functional polymers were first prepared by catalytic chain transfer polymerization (CCTP) using oligo(ethylene glycol) methacrylate as a monomer. The presence of vinylic end groups was then exploited to attach the polymers to thiol functionalized TiO2 nanoparticles via thiol–ene Michael nucleophilic reactions. X-ray photoelectron spectroscopy (XPS), attenuated total reflectance-infrared (ATR-IR), dynamic light scattering (DLS), and thermogravimetric analyses (TGA) were used to verify the successful modification of the TiO2 surface. The modified TiO2 nanoparticles were stable in cell culture media and formed smaller aggregates when compared to non-surface modified nanoparticles. Cellular toxicity of the hybrid TiO2–polymer particles towards human lung cell lines A549 and H1299 in vitro was evaluated. Results from one-dimensional gel electrophoresis show the presence of polymer layers around the particles affects the adsorption of protein onto the TiO2 surface. The reduction in particle aggregate size and changes to the particle surface chemistry, following polymer grafting, was found to reduce cellular uptake and diminish cytotoxicity for both human lung cell lines tested.
Introduction
Hybrid organic–inorganic nanoparticles are of interest for use in various applications due to high surface functionality, afforded by their small size, yielding properties that were substantially different to equivalent bulk materials.1–6 The controlled production of nanoparticles is a growing research area with potential applications in biomedical, optical, and electronic fields.2,5–10 The novel properties of nanomaterials are imbued by large surface effects influencing surface energies, presenting opportunities for functional materials but also in some cases raising concerns over cytotoxicity.11–15 A large number of studies have shown that nanoparticles are able to pass through cell membranes in organisms (leading to their use as drug delivery vehicles),1,6,16 but presently nanoparticle interactions with complex biological systems are still not fully understood.16–18 TiO2 nanoparticles have extended applications in the areas of environmental technologies,19,20 clean energy production,21 self-cleaning surfaces,22 textiles,23 sensors,24 antimicrobial agents, pharmaceuticals, as well as cosmetics, and sunscreens.25,26 The wide-spread use of these particular particles has attracted much research aimed at understanding the biological impact of TiO2 nanoparticles and reducing any toxicity issues.10,13–16,27–29Recently, Tedja et al. showed that the aggregate size of TiO2 nanoparticles in biological media had a significant influence on biological impact of TiO2 on human lung cell lines in vitro (a reduction in TiO2 toxicity was observed when a sonication method was used to reduce the aggregate size30). Micrometer-sized TiO2 aggregates have been found to be more readily taken-up by human lung cell lines when compared to sub-micrometer aggregates; with a corresponding impact on cytotoxicity in the human lung cell lines A549 and H1299. In addition to the particle aggregate size, the surface chemistry of nanoparticles has been shown to affect their biological interactions. Our recent study has shown that adsorption of serum proteins on to the surface of TiO2 nanoparticles resulted in a lower biological impact on both human lung cell lines A549 and H1299.31 Interestingly, the reduction of biological impact due to serum adsorption has been noted to be accompanied by a higher particle uptake compared to the non-serum-treated TiO2 after a 24 h exposure period. Therefore, the adsorption of serum proteins was identified as a potential protection strategy to reduce cytotoxic effects of endocytosed TiO2 nanoparticles.31 However, the adsorption of protein(s) on to nanoparticles results in systems difficult to fully characterize (and therefore control), following the possibility of conformational changes in protein structure and folding. For instance, gold32 and supermagnetic iron oxide33 nanoparticles have been reported to capture serum proteins with significant induced conformational changes. Denatured protein layers may have significant effects on cellular immune responses.34 As a result, strategies to mediate TiO2 nanoparticle surfaces to reduce cytotoxicity whilst minimizing protein adsorption are required.
One practical way to maintain a small aggregate size of nanoparticles is to use a polymer stabilizing outer corona, to increase the steric repulsion between individual nanoparticles. There are several techniques available to coat nanoparticles with polymer, including “grafting to”, “grafting from”, and “layer-by-layer” (LbL) approaches.1,6,35–43 The different synthetic techniques create hybrid organic–inorganic particles with different surface structures. One disadvantage of the LbL approach is that it requires several steps, which can be time consuming and the resulting material can be less stable and sensitive to the ionic concentration, since the surface layers are not covalently bonded.35 Stable polymer coatings may be formed by covalent bonding between the nanoparticle surface and preformed polymers; with either of the grafting approaches.37,38,40 Comparing the two different grafting approaches, the “grafting from” technique usually gives increased grafting density, however subsequent characterization of the polymer layer can be problematic. In contrast, the “grafting to” approach allows for a more facile synthesis and characterization of the polymer prior to nanoparticle surface attachment. In our previous work, we used a variety of functional anchoring groups for the covalent attachment of molecules to the surface of nanoparticles bearing hydroxyl groups, including (for example) dopamine, cysteine, amine, phosphonic acid, carboxylic acid, and trimethoxy silane.40,41,44 A recent study by Tucker-Schwartz et al.45 demonstrated the attachment of a silane compound (that also contained a free thiol) to iron oxide nanoparticles, for subsequent modification via thiol–ene Michael addition.45 In our current work, we used thiol–ene chemistry to modify TiO2 nanoparticles with polymeric chains. The polymer chains were first synthesized by catalytic chain transfer – a polymerization technique ideally suited to making short chains with a very high terminal vinyl functionality for subsequent reaction with thiols.46,47 Thiol–ene addition can be induced easily using two different approaches: thiol–ene Michael nucleophilic addition or photo-initiated radical addition.48–50 After modification and characterization, the TiO2 nanoparticles were assessed in a number of in vitro experiments with the human cell lines A549 and H122.
Materials and methods
Materials
All reagents were used without further purification. The monomer oligo(ethylene glycol) methyl ether methacrylate (Aldrich, 99%) was used as received and stored at −18 °C. The initiator 2,2′ azobisisobutyronitrile (AIBN) was re-crystallized twice from methanol. The catalytic chain transfer polymerization agent bis-(difluoroboryl)dimethylglyoximato cobalt(II) (CoBF) was synthesized according to the method of Bakac et al.51 The solvents acetonitrile (Aldrich, 99%), hexyl amine (Aldrich, 99%), and (3-mercaptopropyl) trimethoxy silane (Aldrich, 98%) were used as received. TiO2 used was Aeroxide-P25 (Evonik, USA) which is a 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 mixture of anatase:rutile with a surface area reported to be approximately 50 m2 g−1. Roswell Park Memorial Institute-1640 (RPMI1640), Dulbecco's modified Eagle medium: nutrient mixture F-12 (DMEM/F-12), defined keratinocyte serum free media (KSFM) cell culture media, fetal bovine serum (FBS), phosphate buffered saline (PBS), Hank's buffered salt solution (HBSS) and Trypsin/EDTA were all purchased from GIBCO Invitrogen. L-glutamine was purchased from Sigma Chemical Co., St. Louis, MO, USA. Plastic culture microplates and flasks used in the experiment were supplied by Sarstedt AG & Co., Germany. CellTiter-Blue® (AlamarBlue®), CellTiter 96® MTS and CellTiter-Glo® Luminescent cell viability assays were purchased from Promega Co., Madison, WI, USA. Hydrochloric acid (HCl) and hydrofluoric acid (HF) were provided from Ajax Finechem Pty Ltd, Sydney, Australia.
:20 mixture of anatase:rutile with a surface area reported to be approximately 50 m2 g−1. Roswell Park Memorial Institute-1640 (RPMI1640), Dulbecco's modified Eagle medium: nutrient mixture F-12 (DMEM/F-12), defined keratinocyte serum free media (KSFM) cell culture media, fetal bovine serum (FBS), phosphate buffered saline (PBS), Hank's buffered salt solution (HBSS) and Trypsin/EDTA were all purchased from GIBCO Invitrogen. L-glutamine was purchased from Sigma Chemical Co., St. Louis, MO, USA. Plastic culture microplates and flasks used in the experiment were supplied by Sarstedt AG & Co., Germany. CellTiter-Blue® (AlamarBlue®), CellTiter 96® MTS and CellTiter-Glo® Luminescent cell viability assays were purchased from Promega Co., Madison, WI, USA. Hydrochloric acid (HCl) and hydrofluoric acid (HF) were provided from Ajax Finechem Pty Ltd, Sydney, Australia.
Cell lines
Human lung epithelial cell lines, A549 (ATCC, CCL-185™) and NCI-H1299 (ATCC, CRL-5803™), kindly provided by Dr Louise Lutze-Mann, were separately cultured in RPMI1640 media supplemented with 10% FBS and 1% L-glutamine. The cells were cultured in treated T75 flasks and incubated at 37 °C in a humidified incubator with 5% CO2 atmosphere (Heraeus). Cell culture experiments were performed in 96-well treated-culture plates for the biological assays (Alamar blue, MTS and ATP luminescent assays) and 6-well treated-culture plates for the cell count and quantification of particle uptake.Synthesis of POEGMA polymer
The CCTP was performed in a similar method to that published elsewhere.52 A typical CCTP procedure is described as follows; the monomer oligo(ethylene glycol) methyl ether methacrylate (OEGMA475) (10 g, 2.1 × 10−2 mol) was mixed with acetonitrile as solvent (10 mL). The reaction mixture was then sparged with N2 for at least an hour. In a different flask, CCTP catalyst CoBF, at a concentration determined by the target molecular weight, was mixed with AIBN (1 mg, 2 × 10−6 mol) and also degassed with N2 to ensure the absence of O2. The reaction solution was then transferred to the flask containing CoBF via a cannula. Polymerization was then run for 14 h at 70 °C and the resulting polymer was purified by precipitation in diethyl ether. The product polymers were characterized using GPC and NMR before use in grafting experiments.Synthesis of TiO2–POEGMA nanoparticles
The synthesis of the nanoparticle–polymer hybrids was done using a two-step process as follows:(1) TiO2 nanoparticles (140 mg, 1.8 × 10−3 mol) were mixed in water (50 mL) with (3-mercaptopropyl) trimethoxysilane (300 mg, 1.5 × 10−3 mol) followed by serial sonication at a frequency of 20 Hz for 10 min intervals (three times with a 5 min break between each). The suspensions were then incubated at 60 °C, with stirring, for 6 h followed by 18 h incubation at room temperature without stirring. The thiolated nanoparticles were purified by a series of washing and centrifugation steps (1 h at 6000 rpm) to ensure the removal of (3-mercaptopropyl) trimethoxysilane.
(2) The thiolated particles were then mixed with a series of POEGMAs with different molecular weights (in acetonitrile and hexylamine). The deoxygenated samples were then placed in a 40 °C oil bath overnight and subsequently purified by centrifugation and freeze-drying.
Polymer and particle characterizations
000000 g mol−1.
Particle uptake measurement
After 24 h incubation with unmodified and modified TiO2 particles at a concentration of 150 μg mL−1, the cell media containing particles was removed and the cells were washed three times with HBSS to remove the non-internalized particles. The cells were then incubated with 1 mL of 6 M HCl overnight. The entire contents of the well were retrieved and each well was washed three times with 1 mL of Milli-Q water. The combined HCl extract together with the washes were treated with 1 mL of 40% (w/w) HF and incubated overnight at room temperature. The samples were then diluted to a final volume of 10 mL with Milli-Q water and the Ti contents were quantified using ICP-OES (PerkinElmer Optima, 3000DV).Biological impact evaluation
The biological impact of nanoparticles was evaluated using three different assays to assess different biological end points: (1) CellTiter-Blue® (AlamarBlue) assay to determine the cell metabolic activity; (2) MTS assay to determine the cell mitochondrial activity; and (3) CellTiter-Glo® luminescent cell viability assay to determine the level of cellular energy (ATP). For each assay, cell cultures were established at an initial cell density of 16700 cells per cm2 and the cells were exposed to a concentration of 150 μg mL−1 of TiO2 for 24 h prior to evaluation.
Statistical analysis
Statistical comparison of multiple groups of data was analysed using one way ANOVA followed by a Dunnett test which was used to compare means from the control group and each of the group exposed to particles. The statistical test was performed using Minitab v13 statistical program (Minitab Inc.). The values are expressed as the mean ± standard error of the mean (SEM). The statistical significance versus control group was established as p < 0.05.Results and discussion
Biocompatible polymer synthesis
The biocompatible polymer chosen for this work was a polyethylene glycol macromonomer analogue made from oligo(ethylene glycol) methyl ether methacrylate (OEGMA) units. OEGMA475 was polymerized using the catalyst chain transfer polymerization (CCTP) technique using bis-(difluoroboryl)dimethylglyoximato cobalt(II) (CoBF), an established and widely used CCT catalyst.51 COBF was shown previously to be highly reactive towards various OEGMA macromonomer radicals with a chain transfer constant (CS) = 1800 for the macromonomer OEGMA475.52In this study, three different polymers with different molecular weights were synthesized via CCTP (Scheme 1) and subsequently characterized by 1H NMR spectroscopy and GPC chromatography (Fig. S1†) – the summary of the polymers obtained is tabulated in Table 1. PDIs of the polymers were obtained via GPC showing PDIs < 1.5 (the actual PDIs from a transfer dominated polymerization should be close or equal to 2, the results obtained herein reflect the inaccurate GPC calibration and losses on purification). Molecular weight values were obtained from both GPC and from 1H NMR data using eqn (1) (below).
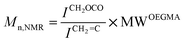 | (1) |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C correspond to the molar masses of OEGMA475, integral of signal at 4.1 ppm (attributed to CH2 in adjacent position of ester group), and of the vinyl group at 5.6–6.0 ppm respectively. Polymer characterization by NMR and GPC were in accord. It is noteworthy that data from the proton NMR (Fig. S1†) can be used to confirm the progress of polymerization and the build up of polymer vinylic end-groups. The polymer vinyl end-groups are distinct from the signals emanating from the monomer vinyl groups (i.e. 5.5 and 6.1 ppm for monomer and 5.6 and 6.0 ppm for polymer respectively).
C correspond to the molar masses of OEGMA475, integral of signal at 4.1 ppm (attributed to CH2 in adjacent position of ester group), and of the vinyl group at 5.6–6.0 ppm respectively. Polymer characterization by NMR and GPC were in accord. It is noteworthy that data from the proton NMR (Fig. S1†) can be used to confirm the progress of polymerization and the build up of polymer vinylic end-groups. The polymer vinyl end-groups are distinct from the signals emanating from the monomer vinyl groups (i.e. 5.5 and 6.1 ppm for monomer and 5.6 and 6.0 ppm for polymer respectively).
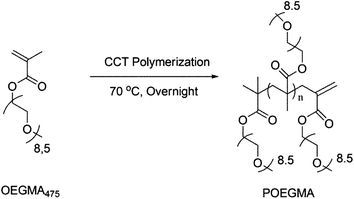 | ||
| Scheme 1 Catalytic chain transfer polymerization of OEGMA yielding vinylic terminated POEGMA. | ||
Synthesis of polymer-coated TiO2 nanoparticles
In this work, surface modification of TiO2 nanoparticles was targeted to; (i) minimize nanoparticle aggregation (on dispersion), and (ii) introduce surface functionality for subsequent attachment to biocompatible polymers. The synthesis of the nanoparticle–polymer hybrids was achieved using a two-step sonochemical and thiol–ene Michael addition process. Firstly, TiO2 nanoparticles were mixed with (3-mercaptopropyl) trimethoxysilane, as a linker with the silyl group attached to TiO2 surfaces leaving exposed free thiol groups (–SH) on the nanoparticle outer surface. The free thiols were then reacted with pre-synthesized polymers using thiol–ene Michael addition reactions as shown in Scheme 2.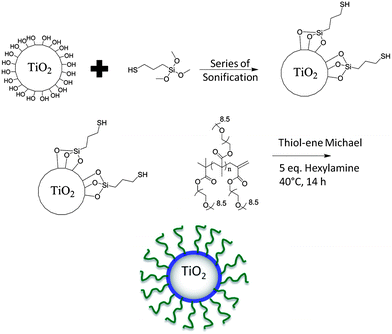 | ||
| Scheme 2 Overall synthetic approach for the surface modification of TiO2 nanoparticles with POEGMA. | ||
In order to ascertain reaction conditions for successful thiol–ene Michael addition reactions, a model reaction was first attempted using the monomer OEGMA475 and the dimer of OEGMA475 (to represent the polymer with vinylic end group) in a reaction with (3-mercaptopropyl) trimethoxysilane catalyzed by hexylamine in acetonitrile. The reaction was followed by ESI-MS mass spectral analysis (as shown in Fig. S2†). It is evident from the mass spectrometry data that on reaction, a shift to higher molecular weights is observed with the m/z peaks increased by 196.4 daltons consistent with the molecular weight of the organo-silane compound. This model reaction confirmed that the reaction of POEGMA (synthesized via CCTP) with 3-mercaptopropyl trimethoxysilane proceeded in a quantitative fashion and (hopefully) could be translated to surface modification (Scheme 2). It is important to note that an attempt was made to attach the resulting polymer bearing silane groups to the surface of nano-TiO2. However, there were difficulties with attachment, as the resulting polymer readily cross-linked forming an insoluble gel network following self-condensation of the terminal silane groups under basic conditions. Hence, the approach shown in Scheme 2, where 3-mercaptopropyl trimethoxysilane was firstly attached to the TiO2 surface was chosen as the preferred synthetic approach. The polymer components (synthesized via CCTP) could be made with a range of different functional monomers (potentially increasing the applicability of the CCT approach). While there has been some work reported in the literature, showing modification of polymers made via CCTP with small thiol bearing molecules,47,52 this is the first time (to our knowledge) that CCT/thiol–ene has been applied to the surface modification of nanoparticles. This synthetic route to PEGylated nanoparticle surfaces provides an alternative to linear PEG with the advantage of significant control over the nature of the PEG surface (possible expansion to copolymerization with control over molecular weight and architecture).53,54
The attachment of linker and polymer were monitored by XPS (Fig. 1). A wide scan survey of the nanoparticle for all elements showed clearly that the bare nano-TiO2 has only Ti and O as the main signals, with slight contamination of carbon from the atmosphere evident (Fig. S3†). After modification with 3-mercaptopropyl trimethoxysilane (the “linker”) and POEGMA there are new elemental signals corresponding to Si, S, O, and C (Fig. S3†), showing successful modification. However, differences reflecting the successive attachment steps involving silane and thiol–ene Michael addition could not be detected by the XPS wide scan. Therefore, high-resolution XPS analysis was carried out to investigate the chemical binding state of each element. XPS evaluation of the different stages of particle functionalization is shown in Fig. 1. XPS of bare TiO2 indicates carbon traces (with a low intensity) attributed to surface contamination by organic compounds during the synthesis or storage. From XPS measurements, after modification with the linker, the particle surface displays signals for C–C or C–H bonds and after attachment of the POEGMA, the XPS spectra confirms the presence of C–O and O–CO bonds in addition to C–C and C–H peaks – clear evidence for the attachment of polymer to the nanoparticle surface. The ratio of C/Ti was also calculated for the nano-TiO2 during different stages of the nanoparticle functionalization. The C/Ti ratio for bare TiO2 is only 0.4 compared to 10.1 after attachment of the linker and 13.5 after attachment of the POEGMA consistent with the successful attachment of linker and subsequent modification with POEGMA.
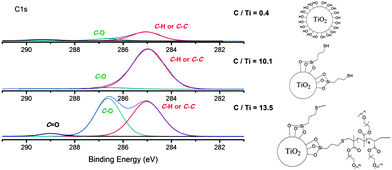 | ||
| Fig. 1 XPS spectra focused on the C1s binding energy of the TiO2–polymer hybrid during different steps of the synthetic process high-resolution spectrum for C1s signal. The C/Ti ratio is indicated in this figure. | ||
The XPS data were further supported by ATR-FTIR analysis (Fig. S4†). It can be seen that the bare TiO2 does not have any signals consistent with organic species except for the broad absorption in the range 3000–3500 cm−1 corresponding to O–H peaks, while analysis of the purified POEGMA modified TiO2 gave characteristic signals at around 1000–1200 cm−1, at 1730 cm−1 and at 2700–2800 cm−1 corresponding to ether bonds (C–O), carbonyl esters (CO) and C–H (aliphatic). The ATR-FTIR results are in good accord with the XPS data.
The synthesized hybrid particles were also characterized by TGA (Fig. S5†) to assess the amount of organic material, viz., OEGMA475 and POEGMA, attached to the TiO2. As expected, TiO2 was stable up to 800 °C. In contrast, those nanoparticles modified with polymer underwent a partial degradation at approximately 330 °C, characteristic of the thermal degradation of POEGMA. The synthesized hybrid nanoparticles underwent thermal weight losses of 12–15%, with the loss slightly higher as the molecular weight of the polymer increased. The grafting density was calculated using the following equation: grafted per nm2 = (ΔMPolymer* × NA)/(MPolymern × mTiO2 × S), with ΔMPolymer*, MPolymern, NA and S corresponding to the mass of polymer grafted (without silane), molecular weight of polymer, Avogadro's number, mass of TiO2 nanoparticles and surface area of TiO2, respectively (Table 2). The number of grafted chains decreased as the molecular weight of the polymer increased. This may be attributed to steric hindrance effects on the thiol–ene Michael additions as the polymer molecular weight increased. As the grafting density is lower than 0.10 chain per nm2 for POEGMA16.0 and 20.0, this indicates that the polymer attached on the surface has a mushroom type conformation. For short OEGMA chain, i.e. 475 g mol−1, the conformation of the polymer attached on TiO2 corresponds to a brush polymer. In summary, the XPS, ATR-IR and TGA analyses were consistent in showing that the polymer modified particles were successfully synthesized.
| Samples | M n (g mol−1) | Weight loss (wt%) | Grafting density (chains per nm2) |
|---|---|---|---|
| a Note: grafting density = (ΔMPolymer × NA)/(MPolymern × mTiO2 × STiO2), with ΔMPolymer, MPolymern, NA and S correspond to mass of polymer grafted determined by TGA, molecular weight of polymer, Avogadro's number and surface area of TiO2, respectively. The grafting density was calculated using 50 m2 g−1 as surface specific). | |||
| TiO2–silane | 196 | 6.0 | — |
| TiO2–OEGMA | 480 | 12 | 1.25 |
| TiO2–POEGMA2.5 | 2600 | 13.5 | 0.39 |
| TiO2–POEGMA16.0 | 16000 |
14 | 0.07 |
| TiO2–POEGMA20.0 | 20000 |
15 | 0.064 |
Finally the polymer-coated nanoparticles were characterized by DLS to investigate particle aggregate sizes in aqueous and cell culture media RPMI1640 supplemented with 10% FBS and 1% L-glutamine. The cell culture media we used has been commonly used for in vitro cell culture systems to support cell growth and proliferation for evaluating material toxicity. In this study, the particle exposure to the cell lines was conducted in this cell culture media; and therefore, it is important to carefully characterize the particles once suspended in this media. Fig. 2 shows that the surface modification of TiO2 nanoparticles by POEGMA-grafting altered the subsequent particle aggregation significantly. The bare TiO2 nanoparticles exhibit aggregation sizes of above 1 μm in all biological media tested in this study. After modification with the linker (silane), the particle aggregate sizes were similar to the bare TiO2. This minimal effect of silane on aggregation may be partly explained by the potential reactivity of the free surface thiols towards disulphide formation. Subsequent attachment of the PEG monomer had no effect on nanoparticle aggregation in the cell culture medium; however, in contrast, attachment of PEG monomer significantly reduced aggregation in water (Fig. 2). Once the TiO2–monomer particles were diluted using the cell culture medium, the particle size aggregate increased to almost 1 μm. This result suggests that the attachment of PEG monomer alone does not confer enough steric stabilization to avoid the aggregation of the nanoparticles in cell culture media. After the attachment of POEGMA, the hybrid particles were successfully stabilized with a particle size of approximately 300 nm (Fig. 2) in the cell culture media (with and without 10% FBS). The data suggests that changing the polymer chain length (between 2.5; 16.0; and 20.000 g mol−1) does not significantly affect the aggregation state of POEGMA–TiO2 particles. A similar observation has been reported with PEG-coated nanospheres designed for drug delivery.55 DLS measurements were also done in various other cell culture media (Fig. S6†), including Dulbecco's modified Eagle medium (DMEM) and keratinocyte serum-free medium (KSFM). The data reported herein is consistent with stabilization yielding a similar aggregate size of 300 nm in all cell culture media tested. The particle stability was monitored over a 24 h period revealing that the POEGMA–TiO2 particles maintained stability with a steady aggregate size of 300 nm (Fig. S7†).
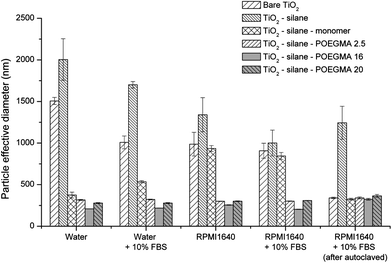 | ||
| Fig. 2 Aggregate sizes of naked and modified TiO2 nanoparticles in different diluents (data taken from at least 5 independent experiments). | ||
Following the modification process, the particles were sterilized by an autoclaving process (121 °C, 1 atm, 20 minutes) and stored at 4 °C for 16 h prior to subsequent biological testing. The DLS measurement shown in Fig. 2 indicates that the aggregate sizes were unaffected by the autoclaving process. Interestingly, the autoclaved bare TiO2 nanoparticles suspended in PBS buffer, (involving a high temperature (121 °C) and storage at 4 °C for 15–16 h), resulted in aggregate sizes significantly smaller than aggregates that were not subjected to an autoclaving process. This suggests that the autoclaving process might have altered the surface of bare TiO2 yielding different interactions with the proteins when suspended in the RPMI1640 cell culture medium supplemented with 10% FBS and 1% L-glutamine. This autoclaving-induced stability effect was not observed for other modified particles.
Cellular uptake of polymer-modified TiO2 nanoparticles by human lung cell lines in vitro
The extent of particle uptake by the cells is shown in Fig. 3 clearly showing that the surface chemistry modifications by silane, as a linker, followed by attachment of POEGMA monomer or polymer significantly affected the amount of particle uptake by both the human lung cell lines A549 and H1299. Fig. 3 shows the amount of particle uptake per cell after a 24 h period of exposure to the naked, silane-, monomer- or polymer-modified TiO2 particles after the sterilization process (autoclaving) at an exposure concentration of 150 μg mL−1. There is a significant decrease of up to 4-fold and 2-fold, of the uptake, after the particles have been modified with silane, by human lung cell lines A549 and H1299, respectively. In our previous investigation, TiO2 nanoparticles with the same surface chemistry comprised of larger particle aggregates were reported to be more readily taken up by cells, compared to smaller particle aggregates.30 In the present study, an increase in particle aggregate size (following attachment of silane, which theoretically made the surface negatively charged), was found to result in a lower cellular uptake, indicating that the surface chemistry plays an important role alongside aggregate size in determining interactions (uptake) with human lung cell lines.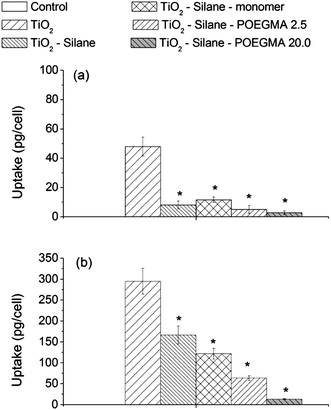 | ||
| Fig. 3 Cellular uptake of naked and modified particles by human lung cell lines (a) A549 and (b) H1299 after a 24 h exposure at a concentration of 150 μg mL−1 TiO2. The * shows the cellular uptake of modified TiO2 that was significantly different from the amount of cellular uptake of the naked TiO2 (by one-way ANOVA followed by Dunnett's test) with p < 0.05, from at least 3 independent experiments. | ||
Modification with OEGMA (monomer) and POEGMA with various chain lengths, resulted in a further reduction in nanoparticle cellular uptake (Fig. 3). This difference in uptake may result from the stability of the particle aggregates; requiring, a longer time period to access the adhered cells on the bottom of the culture dish. The gravitational settling rate of nanoparticles has been reported to significantly affect the amount of internalized particles in the cell culture system.56 Our data shows that particle uptake was reduced by all of the surface modifications studied here. Similar to this finding, PEG-modified polyaspartamide nanoparticles have been shown to escape from being phagocytozed by the mouse monocyte macrophage cell line J774A1,57 thereby minimizing the particle internalization. Conversely, an earlier investigation has reported that the attachment of PEG molecules onto iron oxide nanoparticles significantly increased the particle uptake by the human breast cancer cell line BT20.58
The data in Fig. 3 indicates a reduced particle uptake by A549 cells following silane modification of the nanoparticles and a similar decrease in the particle uptake on further addition with monomer or polymers, while a different extent of cellular uptake reduction is shown for H1299 with the various surface modifications. This result might be attributed to different TiO2 nanoparticle uptake pathways between the two cell lines. A549 and H1299 cell lines have been reported to internalize TiO2 nanoparticles via different active uptake endocytosis pathways; clathrin-dependent and clathrin-/caveolae-independent endocytosis, respectively.31 Another possibility is that a very low uptake for silane-modified particles by A549 cells was minimally affected by further modifications.
Protein adsorption profiles of naked and modified nanoparticles
Fig. 4 shows the SDS-PAGE of proteins attached to the naked and modified TiO2 nanoparticles following a 24 h period of incubation in RPMI1640 cell culture media supplemented with 10% fetal bovine serum (FBS) and 1% L-glutamine at 37 °C. Our previous study has shown that the amount of particle cellular uptake was directly affected by the quantity and identity of protein(s) adsorbed onto the surface of nanoparticles.31 From the SDS-PAGE gel, the bare TiO2 particles are shown to adsorb at least 15 different proteins, as shown by the number of bands in lane 2 (Fig. 4).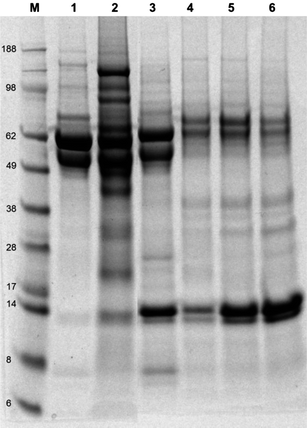 | ||
| Fig. 4 SDS-PAGE electrophoresis gel of proteins adsorbed onto the surface of non- and modified-TiO2 nanoparticles after suspension in RPMI1640 supplemented with 10% FBS and 1% L-glutamine for an overnight period at 37 °C, line M: see Blue 2 marker, line 1: FBS, line 2: TiO2, line 3: TiO2–silane, line 4: TiO2–silane–monomer, line 5: TiO2–silane–POEGMA 2500 g mol−1, line 6: TiO2–silane–POEGMA 20000 g mol−1. | ||
Fig. 4 shows the profile of adsorbed protein onto the surface of TiO2 nanoparticles analyzed by protein (SDS-PAGE) gel electrophoresis. TiO2–silane particles adsorbed fewer proteins, as some of the proteins (in particular with high molecular weights) that adsorb on the naked nanoparticle sample were no longer adsorbed onto the surface of TiO2–silane particles. Furthermore, the results show that the low molecular weight proteins (approximately 14 kDa) appeared to be preferentially adsorbed onto TiO2–silane particles, while the higher molecular weight proteins were not observed. Further modification with either monomer OEGMA475 or polymer POEGMA with different chain lengths significantly changed the adsorbed protein profile and a reduction in the quantity of protein adsorbed (visualized by further reduction of the number of bands). The band associated with the adsorption of proteins in the low molecular weight range is shown to be more intense as the chain length of OEGMA increases, reflecting increased protein adsorption capacity of proteins in the low molecular weight range.
There are a number of possible low molecular weight serum-derived proteins that may be adsorbed onto the surface of TiO2 nanoparticles, such as (but not limited to): transthyretin, haptoglobin α1 chain, lysozyme C, and apolipoproteins (ApoC-III, ApoC-II, and ApoA-II). Transthyretin is a thyroxin transport protein that is known to adsorb to toxic components present in the blood stream.59 Transthyretin specifically interacts with receptor-associated proteins in the liver (it's major site of degradation) and has been reported to be strongly associated with silica nanoparticles.60 Haptoglobin α1 chain is a protein known to interact with free hemoglobin (Hb) to prevent oxidative damage by Hb, and is also known to promote receptor recognition by monocytes/macrophages. Another protein that may be attached to our modified particles is lysozyme C, known to interact with TiO2 nanoparticles.61 The adsorption of one or more of these proteins may have a significant impact how the cell interacts with the nanoparticles. Interestingly, the modification of TiO2 nanoparticles with monomer OEGMA475 or POEGMA is shown to significantly reduce the adsorption of protein(s) with molecular weights in the range of approximately 58 and 98 kDa. These protein bands include vitronectin (shown in our previous study), associated with an increase in the amount of particle taken up by human lung cell line A549.31 Vitronectin has also been demonstrated to increase crocidolite asbestos uptake by rabbit pleural mesothelial cells via integrin receptors.62 Therefore, it is possible that the reduction of particle cellular uptake of modified particles can be attributed to a reduction in adsorbed vitronectin.
Biological impact of the hybrid nanoparticles on human lung cell lines in vitro
Fig. 5 shows the biological impact of non- and modified-TiO2 nanoparticles based on cellular metabolic and mitochondrial activities, and also cellular ATP level following 24 h exposure at a concentration of 150 μg mL−1 TiO2 nanoparticles. This particle concentration was particularly chosen as it was the concentration at which naked TiO2 started showing biological impact on human lung cell lines in vitro.30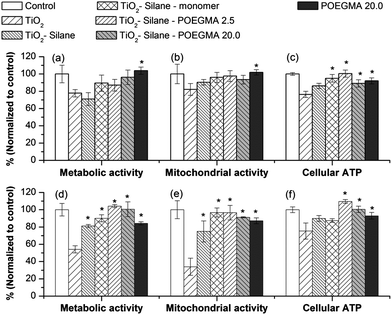 | ||
| Fig. 5 Biological impact of TiO2 nanoparticles before and after modification in human lung cell lines (a–c) A549 and (d–f) H1299 after a 24 h exposure at a concentration of 150 μg mL−1 TiO2. The * shows the biological impact of modified TiO2 that was significantly different from the amount of cellular uptake of the naked TiO2 (by one-way ANOVA followed by Dunnett's test) with p < 0.05, from at least 4 independent experiments. | ||
The data in Fig. 5, shows that modification of the nanoparticles with POEGMA polymer reduces the biological impact of TiO2 particles especially in the case of H1299, which is more sensitive to particle exposure compared to A549. The sensitivity of H1299 has also been noted in previous investigations.30,31 Modification with silane alone also reduced the biological impact of TiO2 nanoparticles. This could be due to the reduction in protein adsorbed onto the silane-modified particles compared to the naked (unmodified) particles, resulting in significantly less particle cellular uptake.
Regardless of the different chain lengths of POEGMA (Fig. 5), the functionalization of the nanoparticles improved biocompatibility. This improvement in the biocompatibility of TiO2 nanoparticles correlates with a lower particle uptake by both human lung cell lines A549 and H1299, which may be directly linked with a reduction in the quantity and different type of proteins adsorbed onto the particle surface. In our previous investigation, the adsorption of proteins (by pre-exposure to FBS) was shown to increase the particle uptake, while interestingly resulting in a lower biological impact after a 24 h exposure.31 The data from this study shows that modifying nanoparticle surface chemistry also influences the spectrum of proteins binding, suggesting that the type and amount of proteins binding to the particle surface, mediated by altering particle surface chemistry, is very important in determining how the particles interact with cells with a consequential impact on cell health.
Conclusion
In this work, we report a facile method to modify TiO2 nanoparticles using thiol–ene click chemistry and CCT polymerization. The synthetic method described herein could be broadened to attach polymers onto a range of oxide type nanoparticles. The attachment of POEGMA to nano-TiO2 aided in maintaining stable aggregates in various aqueous and biological media. The surface modifications were also shown to change the type and amount of serum proteins adsorbed onto the nano-TiO2. The surface chemistry was shown to play an important role in determining the amount of particle uptake by cells (in addition to the aggregate sizes). It is noteworthy that the adsorption of vitronectin, a protein that has been highlighted to trigger TiO2 cellular uptake, was reduced following polymer modification and this may therefore contribute to a lower cellular uptake of particles, especially for the A549 cell line. A reduced nanoparticle cell uptake leads to a demonstrated decrease in the biological impact of the TiO2 on the two different human lung cell lines A549 and H1299. The results presented in this work have important implications for potentially addressing/investigating concerns regarding the nanotoxicity of nano-TiO2 materials.Acknowledgements
The authors acknowledge the Australian Research Council (ARC) for funding. CB is thankful for his APD fellowship from ARC (DP 1092640).References
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- P. Alivisatos, Nat. Biotechnol., 2004, 22, 47–52 CrossRef CAS.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS.
- A. Balazs, T. Emrick and T. Russell, Science, 2006, 314, 1107–1110 CrossRef CAS.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS.
- M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225–4241 CrossRef CAS.
- P. Katangur, P. K. Patra and S. B. Warner, Polym. Degrad. Stab., 2006, 91, 2437–2442 CrossRef CAS.
- K. M. L. Taylor-Pashow, J. Della Rocca, R. C. Huxford and W. Lin, Chem. Commun., 2010, 46, 5832–5849 RSC.
- L. Yan, Z. Yu, L. Chen, C. Wang and S. Chen, Langmuir, 2010, 26, 10657–10662 CrossRef CAS.
- H. F. Krug and P. Wick, Angew. Chem., Int. Ed., 2011, 50, 1260–1278 CrossRef CAS.
- M. C. Hersam, N. P. Guisinger and J. W. Lyding, Nanotechnology, 2000, 11, 70–76 CrossRef CAS.
- M. Anisa, S. D. Abdallah and A. S. Peter, Nanotechnology, 2003, 14, R9–R13 CrossRef.
- P. O. Andersson, C. Lejon, B. Ekstrand-Hammarström, C. Akfur, L. Ahlinder, A. Bucht and L. Österlund, Small, 2011, 7, 514–523 CrossRef CAS.
- Y.-F. Li and C. Chen, Small, 2011, 7, 2965–2980 CrossRef CAS.
- K. Cai, Y. Hou, Y. Hu, L. Zhao, Z. Luo, Y. Shi, M. Lai, W. Yang and P. Liu, Small, 2011, 7, 3026–3031 CrossRef CAS.
- Z. Pan, W. Lee, L. Slutsky, R. A. F. Clark, N. Pernodet and M. H. Rafailovich, Small, 2009, 5, 511–520 CrossRef CAS.
- J. Panyam and V. Labhasetwar, Pharm. Res., 2003, 20, 212–220 CrossRef CAS.
- K. T. Thurn, H. Arora, T. Paunesku, A. Wu, E. Brown, C. Doty, J. Kremer and G. Woloschak, Nanomedicine, 2011, 7, 123–130 CrossRef CAS.
- S. Liu, M. Lim, R. Fabris, C. Chow, M. Drikas and R. Amal, Environ. Sci. Technol., 2008, 42, 6218–6223 CrossRef CAS.
- (a) C. Young, T. M. Lim, K. Chiang, J. Scott and R. Amal, Appl. Catal., B, 2008, 78, 1–10 CrossRef CAS; (b) Z. R. Paz, Beilstein J. Nanotechnol., 2011, 2, 845–861 CrossRef.
- Y. K. Kho, A. Iwase, W. Y. Teoh, L. Mädler, A. Kudo and R. Amal, J. Phys. Chem. C, 2010, 114, 2821–2829 CAS.
- S. W. Lam, A. Soetanto and R. Amal, J. Nanopart. Res., 2009, 11, 1971–1979 CrossRef CAS.
- C. Gunawan, W. Teoh, C. Marquis, J. Lifia and R. Amal, Small, 2009, 5, 341–344 CrossRef CAS.
- M.-I. Baraton and L. Merhari, J. Eur. Ceram. Soc., 2004, 24, 1399–1404 CrossRef CAS.
- G. J. Nohynek, J. Lademann, C. Ribaud and M. S. Roberts, Crit. Rev. Toxicol., 2007, 37, 251–277 CrossRef CAS.
- O. Carp, C. L. Huisman and A. Reller, Prog. Solid State Chem., 2004, 32, 33–177 CrossRef CAS.
- D. R. Sambandan and D. Ratner, J. Am. Acad. Dermatol., 2011, 64, 748–758 CrossRef CAS.
- V. H. Grassian, A. Adamcakova-Dodd, J. M. Pettibone, P. I. O'shaughnessy and P. S. Thorne, Nanotoxicology, 2007, 1, 211–226 CrossRef CAS.
- G. Roebben, S. Ramirez-Garcia, V. Hackley, M. Roesslein, F. Klaessig, V. Kestens, I. Lynch, C. Garner, A. Rawle, A. Elder, V. Colvin, W. Kreyling, H. Krug, Z. Lewicka, S. McNeil, A. Nel, A. Patri, P. Wick, M. Wiesner, T. Xia, G. Oberdörster and K. Dawson, J. Nanopart. Res., 2011, 13, 2675–2687 CrossRef.
- R. Tedja, C. Marquis, M. Lim and R. Amal, J. Nanopart. Res., 2011, 13, 3801–3813 CrossRef CAS.
- R. Tedja, M. Lim, R. Amal and C. Marquis, ACS Nano, 2012, 6, 4083–4093 CrossRef CAS.
- S. H. D. P. Lacerda, J. J. Park, C. Meuse, D. Pristinski, M. L. Becker, A. Karim and J. F. Douglas, ACS Nano, 2009, 4, 365–379 CrossRef.
- (a) M. Mahmoudi, M. A. Shokrgozar, S. Sardari, M. K. Moghadam, H. Vali, S. Laurent and P. Stroeve, Nanoscale, 2011, 3, 1127–1138 RSC; (b) P. Maffre, K. Nienghaus, F. Amin, W. J. Parak and G. U. Nienhaus, Beilstein J. Nanotechnol., 2011, 2, 347–383 CrossRef.
- S. J. Soenen, P. Rivera-Gil, J. M. Montenegro, W. J. Parak, S. C. De Smedt and K. Braeckmans, Nano Today, 2011, 6, 446–465 CrossRef CAS.
- G. Schneider and G. Decher, Nano Lett., 2004, 4, 1833–1839 CrossRef CAS.
- A. Reisch, J. Hemmerle, J.-C. Voegel, E. Gonthier, G. Decher, N. Benkirane-Jessel, A. Chassepot, D. Mertz, P. Lavalle, P. Mesini and P. Schaaf, J. Mater. Chem., 2008, 18, 4242–4245 RSC.
- Z. Lu, J. Wang, Q. Li, L. Chen and S. Chen, Eur. Polym. J., 2009, 45, 1072–1079 CrossRef CAS.
- (a) C. Boyer, V. Bulmus, P. Priyanto, W. Y. Teoh, R. Amal and T. P. Davis, J. Mater. Chem., 2009, 19, 111–123 RSC; (b) C. Boyer, M. R. Whittaker, M. Luzon and T. P. Davis, Macromolecules, 2009, 42, 6917–6926 CrossRef CAS.
- Y. Q. Wang, G. P. Zeng, T. R. Sun, X. L. Hong, J. X. Ling and G. Y. Zhang, e-Polym., 2010, 112 Search PubMed.
- C. Boyer, P. Priyanto, T. P. Davis, D. Pissuwan, V. Bulmus, M. Kavallaris, W. Y. Teoh, R. Amal, M. Carroll, R. Woodward and T. St Pierre, J. Mater. Chem., 2010, 20, 255–265 RSC.
- C. Boyer, M. R. Whittaker, V. Bulmus, J. Liu and T. P. Davis, NPG Asia Mater., 2010, 2, 23–30 CrossRef.
- A. B. Lowe, B. S. Sumerlin, M. S. Donovan and C. L. McCormick, J. Am. Chem. Soc., 2002, 124, 11562–11563 CrossRef CAS.
- P. S. Ghosh, C.-K. Kim, G. Han, N. S. Forbes and V. M. Rotello, ACS Nano, 2008, 2, 2213–2218 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. B. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS.
- A. K. Tucker-Schwartz, R. A. Farrell and R. L. Garrell, J. Am. Chem. Soc., 2011, 133, 11026–11029 CrossRef CAS.
- (a) M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS; (b) P. J. Roth, C. Boyer, A. B. Lowe and T. P. Davis, Macromol. Rapid Commun., 2011, 32, 1123–1143 CrossRef CAS; (c) N. B. Cramer, J. P. Scott and C. N. Bowman, Macromolecules, 2002, 35, 5361–5365 CrossRef CAS; (d) C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS; (e) A. B. Lowe, Polym. Chem., 2010, 1, 17–38 RSC; (f) S. P. S. Koo, M. M. Stamenović, R. A. Prasath, A. J. Inglis, F. E. Du Prez, C. Barner-Kowollik, W. Van Camp and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1699 CrossRef CAS; (g) C. Boyer, J. Liu, V. Bulmus and T. P. Davis, Aust. J. Chem., 2009, 62(8), 830 CrossRef CAS; (h) H. T. Ho, M. E. Levere, S. Pascual, V. Montembault, J.-C. Soutif and L. Fontaine, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1657 CrossRef CAS; (i) C. Boyer and T. P. Davis, Chem. Commun., 2009, 6029 RSC; (j) C. Boyer, V. Bulmus and T. P. Davis, Macromol. Rapid Commun., 2009, 30, 493 CrossRef CAS; (k) M. Li, P. De, H. Li and B. S. Sumerlin, Polym. Chem., 2010, 1, 854 RSC.
- G.-Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196–1204 RSC.
- M. Uygun, M. A. Tasdelen and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 103–110 CrossRef CAS.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- A. Bakac, M. E. Brynildson and J. H. Espenson, Inorg. Chem., 1986, 25, 4108–4114 CrossRef CAS.
- (a) J. P. A. Heuts and N. M. B. Smeets, Polym. Chem., 2011, 2, 2407–2423 RSC; (b) G. C. Sanders, B. G. P. van Ravensteijn, R. Duchateau and J. P. A. Heuts, Polym. Chem., 2012, 3, 2200–2208 RSC; (c) T. Y. J. Chiu, J. P. A. Heuts, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Chem. Phys., 2004, 205, 752 CrossRef CAS; (d) Q. Zhang, S. Slavin, M. W. Jones, A. J. Haddleton and D. M. Haddleton, Polym. Chem., 2012, 3, 1016–1023 RSC; (e) D. M. Haddleton, D. R. Morsley, J. P. O'Donnell and S. N. Richards, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 3549 CrossRef CAS; (f) K. A. McEwan and D. M. Haddleton, Polym. Chem., 2011, 2, 1992–1999 RSC; (g) N. M. B. Smeets, T. G. T. Jansen, J. P. A. Heuts, A. M. van Herk and J. Meuldijk, Macromol. React. Eng., 2012, 6, 110–118 CrossRef CAS.
- (a) A. H. Soeriyadi, G.-Z. Li, S. Slavin, M. W. Jones, C. M. Amos, C. R. Becer, M. R. Whittaker, D. M. Haddleton, C. Boyer and T. P. Davis, Polym. Chem., 2011, 2, 815–822 RSC; (b) S. Slavin, E. Khoshdel and D. M. Haddleton, Polym. Chem., 2012, 3, 1461–1466 RSC; (c) L. Nurmi, J. Lindqvist, R. Randev, J. Syrett and D. M. Haddleton, Chem. Commun., 2009, 2727–2729 RSC; (d) J. Mazzolini, O. Boyron, V. Monteil, F. D'Agosto, C. Boisson, G. C. Sanders, J. P. A. Heuts, R. Duchateau, D. Gigmes and D. Bertin, Polym. Chem., 2012 10.1039/c2py20199b.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773–3794 CrossRef CAS.
- M. T. Peracchia, R. Gref, Y. Minamitake, A. Domb, N. Lotan and R. Langer, J. Controlled Release, 1997, 46, 223–231 CrossRef CAS.
- K. Wittmaack, ACS Nano, 2011, 5, 3766–3778 CrossRef CAS.
- E. F. Craparo, G. Cavallaro, M. L. Bondi, D. Mandracchia and G. Giammona, Biomacromolecules, 2006, 7, 3083–3092 CrossRef CAS.
- Y. Zhang, N. Kohler and M. Zhang, Biomaterials, 2002, 23, 1553–1561 CrossRef CAS.
- T. Hamers, J. H. Kamstra, P. H. Cenijn, K. Pencikova, L. Palkova, P. Simeckova, J. Vondracek, P. L. Andersson, M. Stenberg and M. Machala, Toxicol. Sci., 2011, 121, 88–100 CrossRef CAS.
- Y. M. Kim, S. I. Chung and S. Y. Lee, Toxicol. Lett., 2005, 158, 1–9 CrossRef CAS.
- Z. Xu, X. W. Liu, Y. S. Ma and H. W. Gao, Environ. Sci. Pollut. Res., 2010, 17, 798–806 CrossRef CAS.
- A. M. Boylan, D. A. Sanan, D. Sheppard and V. C. Broaddus, J. Clin. Invest., 1995, 96, 1987–2001 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental information, Fig. S1–S7 and Table S1 are available. See DOI: 10.1039/c2py20450a |
| This journal is © The Royal Society of Chemistry 2012 |