Synthesis of a poly(2-azanorbornene) with a high degree of cis-TT-stereoregularity and a regular secondary solution structure†
Elisabeth
Rossegger
a,
László
Oláh
a,
Roland
Fischer
b,
Petra
Kaschnitz
c,
Olívia
Varga
d,
Mihály
Kállay
d,
Gregor
Scheipl
e,
Franz
Stelzer
c and
Frank
Wiesbrock
*c
aPolymer Competence Center Leoben GmbH (PCCL), Roseggerstraße 12, 8700 Leoben, Austria
bGraz University of Technology, Institute of Inorganic Chemistry, Stremayrgasse 9, 8010 Graz, Austria
cGraz University of Technology, Institute for Chemistry and Technology of Materials, Stremayrgasse 9, 8010 Graz, Austria. E-mail: f.wiesbrock@tugraz.at; Fax: +43 316 873 10 32283; Tel: +43 316 873 32283
dBudapest University of Technology and Economics (BME), Department of Physical Chemistry and Materials Science, P.O. Box 91, 1521 Budapest, Hungary
eJoanneum Research, Materials Micro- and Nanostructuring, Leonhardstrasse 59, 8010 Graz, Austria
First published on 17th July 2012
Abstract
Methyl-N-(1-phenylethyl)-2-azabicyclo[2.2.1]hept-5-ene-3-carboxylate (AzaN) was synthesized as a diastereomerically pure monomer and subsequently polymerized with the ruthenium-based M31® catalyst. The poly(2-azanorbornene)s with polymerization degrees in the range from 100 to 300 exhibited high degrees of cis-TT stereoregularity in contrast to previously reported poly(2-azanorbornene)s that were prepared with molybdenum-based catalysts and exhibited cis-HT and trans-HT stereoregularity. The formation of regular secondary structures in acetonitrile solution was verified by circular dichroism and AFM measurements and supported by theoretical calculations of a polymer model with eight repeating units. Of special note are the large diameters of the secondary structures that are indicative of a multiple-strand filament.
Introduction
The ring-opening metathesis polymerization (ROMP) is a versatile polymerization technique for the synthesis of highly diverse macromolecules using Mo-based Schrock-type or Ru-based Grubbs-type initiators.1–4 Recent trends and developments in the well-established ROMP technique were the topic of a huge number of publications in the last few years. With the introduction of 3rd generation Grubbs initiators, tremendous progress has been made concerning the precise synthesis of regular olefinic polymers with diverse architectures.5–8Azanorbornenes are optically active nitrogen-containing compounds that are synthetic precursors for a large variety of natural products like amino acids.9,10 Further developments of the [4 + 2] Diels–Alder cycloaddition11 paved the way for the hetero Diels–Alder reaction, which is of great benefit for the stereoselective asymmetric synthesis of heterocycles, including highly functionalized natural products.12–15 A further step in the direction of material design concerns the control of the stereochemistry of polymers obtained by hetero Diels–Alder reactions. Correspondingly, the synthesis of polymers bearing bioactive or electroactive functions within their polymer structure is possible. Recent findings in the understanding of the mechanism and synthesis of organometallic chemistry have paved the way for realizing the synthesis of tailor made complex materials.16 These macromolecules find utilities in applications with targeted mechanical, biological and electronic properties.
However, the polymerization of monomers with amine functionalities still faces challenges due to initiator deactivation, but, concomitant with the introduction of 3rd generation Grubbs initiators, these days state-of-the-art ROMP offers the advantage of paramount functional group tolerance, including tertiary amines.17,18 Hence, the incorporation of highly diverse chemical groups into the targeted materials is greatly facilitated and azanorbornenes are of topical interest for the synthesis of various bio-inspired agents. Studies on the tacticity and stereochemistry in polymers and also norbornene derivatives undergoing metathesis polymerization were published in the literature.19–21 The ability to prepare stereoregular polymers with ROMP appears to be actually limited to Mo- and W-derived catalysts at this time. Ru-based catalysts have not yet shown similar or comparable activity.22,23
Previous work reported the polymerization of methyl-N-(1-phenylethyl)-2-azabicyclo[2.2.1]hept-5-ene-3-carboxylate (AzaN) with a Mo-based metathesis catalyst and an immobilized, heterogenized Mo-catalyst, which yielded different stereoregular structures (Scheme 1).24–26 In this publication, the diastereoselective monomer synthesis of AzaN and its highly stereoselective polymerization using a Ru-based initiator are described. The solution secondary structure of pAzaN was thoroughly investigated by means of circular dichroism and AFM measurements, and supported by modelling studies.
 | ||
| Scheme 1 Schematic representation of the ROM polymerization of AzaN, catalyzed by Ru-based Umicore M31® (top). Examples of stereoselectivity in poly(azanorbornene)s: (a) cis-HT isotacticity (preparable with a homogeneous Mo-based Mo(CH-t-Bu)(NAr)(OC(CH3)(CF3)2)2 catalyst),24,25 (b) trans-HT isotacticity (preparable with a heterogeneous Mo-based catalyst Mo(CH-t-Bu)(NAr)((OR))2 + SiO2/AlOR with R = C(CH3)2CF3 or CCH3(CF3)2),26−27(c) cis-TT syndiotacticity (reported in here for pAzaN), and (d) trans-TT syndiotacticity. | ||
Experimental
Materials
Boron trifluoride diethyl etherate, dichloromethane, cyclohexane, ethyl acetate, (R)-(+)-α-methylbenzylamine (98%), molecular sieves (4 Å), trifluoroacetic acid (99%), and ethyl vinyl ether were purchased from Sigma-Aldrich (Vienna, Austria) and used as received. Dicyclopentadiene from Sigma-Aldrich (Vienna, Austria) was freshly cracked prior to performance of the hetero Diels–Alder reaction. HPLC grade acetonitrile was purchased from Merck (Budapest, Hungary). Methylglyoxylate hemiacetal was used as received from KEKELIT GmbH (Linz, Austria). The catalyst Umicore M31®, [1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro-(3-phenyl-1H-inden-1-ylidene)(pyridyl)ruthenium(II), was supplied by Umicore AG & Co. KG (Hanau, Germany).Instrumentation
FT-IR spectra were recorded with a Perkin Elmer Spectrum One instrument with a Harrick ATR accessory (spectral range 4000–450 cm−1). All NMR spectra were recorded on a Bruker 300 MHz spectrometer with the exception of the NOESY spectra, which were recorded on a Varian Unity Inova 500 MHz NB High Resolution FT NMR. The solvent residual peak of CDCl3 was used for referencing the spectra to 7.26 ppm and 77.16 ppm, respectively. Peak shapes are indicated as follows: s (singlet), bs (broad singlet), d (doublet), dd (doublet of doublets), m (multiplet). Average molecular weights and polydispersity indices were determined by size exclusion chromatography GPC using CHCl3/Et3N/isoPrOH (94/4/2) as eluent. The measurements were performed with a Merck Hitachi L-6000A pump, separation columns from Polymer Standards Service, 8 × 300 mm STV linear XL 5 μm-grade size, and a differential refractometer Waters 410 detector. Polystyrene standards from Polymer Standard Service were used for calibration. Differential scanning calorimetry measurements were made with a Perkin Elmer Pyris Diamond Differential Scanning Calorimeter, equipped with a Perkin Elmer CCA7 cooling system using liquid nitrogen. A nitrogen flow of 20 mL min−1 and heating/cooling rates of 10 K min−1 were used. Crystal data were collected and integrated with a Bruker APEX-II CCD system with monochromated Mo Kα radiation (λ = 0.71073 Å) at 100 K. The structures were solved by direct methods using SHELXS-97 and refined by full matrix least squares calculations on F2 with SHELXL-97.28,29 All protons on carbon atoms were calculated and allowed to ride on their parent atoms with fixed isotropic contributions. Extinction corrections were applied for the compound using SADABS. UV-VIS absorption and circular dichroism spectra were recorded on an Agilent 8453 diode array spectrometer and a Jasco J-810 spectropolarimeter, respectively, in solutions of the monomer and the polymer in HPLC grade acetonitrile at room temperature. Polymer films were spincoated with the Karl Suss CT-62 Spin Coater (20 s, 1000 rpm). AFM images were taken with a Digital Instruments Dimension (TM) 3100 Scanning Probe Microscope (VEECO) with Olympus Cantilevers AC160TS (exhibiting almost symmetrical side angles with respect to the substrate, yielding comparable aspect ratios in all directions of the image), resonance frequency 300 kHz, spring constant 42 N m−1, and a tip radius <10 nm for the highest resolution imaging. Polymer samples for AFM measurements were prepared by spincoating 400 μL of a solution of pAzaN300 (c = 0.04 mg mL−1) onto 1 × 1 cm silicon wafers. Optical rotation was measured on a Perkin Elmer 341 polarimeter at the wavelength of 589 nm and a path length of 10 cm at 20 °C in dichloromethane.Preparation of AzaN
The monomer was synthesized from methylglyoxylate hemiacetal (6.60 g, 55 mmol, 1.06 eq.), (R)-1-methylbenzylamine (7 mL, 58 mmol, 1.12 eq.), trifluoroacetic acid (4 mL, 52 mmol, 1.00 eq.), boron trifluoride etherate (6.4 mL, 52 mmol, 1.00 eq.), and cyclopentadiene (5 mL, 60 mmol, 1.15 eq.) according to a slightly modified literature recipe.24 The reaction temperature was maintained at −60 °C for 7 h in order to optimize the yield of the diastereomerically pure product. The recovered monomer (yield: 3.68 g, 14.30 mmol, 27.5%) was additionally purified by column chromatography (silica gel, CH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc = 50:1). Single crystals of the monomer were obtained from recrystallization in dichloromethane and storage at +4 °C for three days, yield: 87%. FT-IR: 3120–2880 νstr(C–H), 2850–2810 νstr(O–CH3), 1713 νstr(C
:EtOAc = 50:1). Single crystals of the monomer were obtained from recrystallization in dichloromethane and storage at +4 °C for three days, yield: 87%. FT-IR: 3120–2880 νstr(C–H), 2850–2810 νstr(O–CH3), 1713 νstr(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1371 δs(CH3), 1279–1254 νsym(cis –CHCH–), 1230–1198 νstr(C–N), 1189 νstr(O–CH3), 775–755 ν(C–H), 721–682 ν(cis –CHCH–). 1H-NMR (20 °C, CDCl3, 300 MHz): δ (ppm) = 7.40–7.24 (5H, m, Ar–H), 6.43–6.40 (1H, m, H5), 6.04 (1H, m, H6), 3.79 (3H, s, H12), 3.54 (1H, s, H1), 3.10 (1H, s, H4), 3.03 (1H, q, 3JH–H = 6.3 Hz, H8), 2.49 (1H, s, H3), 1.89 (1H, m, H7′), 1.21, 1.23 (4H, s/d, 3JH–H = 6.3 Hz, H7, H9). 13C-NMR (20 °C, CDCl3, 75 MHz): δ (ppm) = 175.5 (C11), 145.1 (C10), 136.0 (C5), 133.9 (C6), 128.5/127.7/127.2 (Ar–C), 64.5 (C3), 63.7 (C8), 63.6 (C1), 52.4 (C12), 49.8 (C4), 45.9 (C7), 23.9 (C9). [α]20D = +113.2° (c = 0.7, CH2Cl2).
O), 1371 δs(CH3), 1279–1254 νsym(cis –CHCH–), 1230–1198 νstr(C–N), 1189 νstr(O–CH3), 775–755 ν(C–H), 721–682 ν(cis –CHCH–). 1H-NMR (20 °C, CDCl3, 300 MHz): δ (ppm) = 7.40–7.24 (5H, m, Ar–H), 6.43–6.40 (1H, m, H5), 6.04 (1H, m, H6), 3.79 (3H, s, H12), 3.54 (1H, s, H1), 3.10 (1H, s, H4), 3.03 (1H, q, 3JH–H = 6.3 Hz, H8), 2.49 (1H, s, H3), 1.89 (1H, m, H7′), 1.21, 1.23 (4H, s/d, 3JH–H = 6.3 Hz, H7, H9). 13C-NMR (20 °C, CDCl3, 75 MHz): δ (ppm) = 175.5 (C11), 145.1 (C10), 136.0 (C5), 133.9 (C6), 128.5/127.7/127.2 (Ar–C), 64.5 (C3), 63.7 (C8), 63.6 (C1), 52.4 (C12), 49.8 (C4), 45.9 (C7), 23.9 (C9). [α]20D = +113.2° (c = 0.7, CH2Cl2).
Crystal data for AzaN
M = 257.33, monoclinic, a = 11.3513(19), b = 5.7944(10), c = 11.730(2) Å, β = 117.303(6)°, space group P21, Z = 2, V = 685.6(2) Å3, μ(Mo-Kα) = 0.082 mm−1, 6920 measured and 2876 unique reflections [R(int) = 0.0567], wR2 = 0.0503, R = 0.0837 for 2876 reflections [I ≥ 2σ(I)] and 174 parameters. The function minimized was wR2 = {Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]}1/2; w = 1/[σ2(Fo2) + (ap)2 + bp]; p = (Fo2 + 2Fc2)/3; a = 0.0358, b = 0. Residual electron density was 0.166/−0.176 Å−3.General synthesis of pAzaNn (n = 100, 150, 200, 300)
The monomer AzaN (200 mg, 0.77 mmol) was dissolved in dichloromethane (6 mL) under an inert atmosphere, and the catalyst Umicore M31® was added.30 Monomer conversion was monitored by thin layer chromatography (CH:EtOAc = 5:1). The polymerization was quenched with ethyl vinyl ether after full monomer conversion. The product was purified by precipitation in cold ethanol from dichloromethane solutions and subsequently dried in vacuum; yield: 70% of a brown solid. The same procedure was carried out for all polymers; however, a second amount of catalyst needed to be added for full monomer conversion of the M:I ratios of 200:1 and 300:1 (Table 1). The glass-transition temperature Tg of the polymers was determined by DSC and found at 89.3 °C; STA analysis showed a mass loss of 5% at 308 °C. FT-IR: 3120–2880 νstr(–C–H), 2850–2810 νstr(O–CH3), 1732 νstr(CO), 1371 δs(–CH3), 1272–1251 νsym(cis –CHCH–), 1200 νstr(O–CH3), 1225–1123 νstr(C–N), 777–731 ν(C–H), 718–676 ν(cis –CHCH–). 1H-NMR (20 °C, CDCl3, 300 MHz): δ (ppm) = 7.39–7.06 (5H, bs, Ar–H), 5.86–5.20 (2H, bs, H6, H5), 4.35–3.75 (2H, H1, H8), 3.64–3.17 (4H, bs, H12, H3), 3.14–2.22 (2H, bs, H4, H7′), 1.51–1.01 (4H, bs, H7, H9). 13C-NMR (20 °C, CDCl3, 75 MHz): δ (ppm) = 174.5 (C11), 145.3 (C10), 136.7 (C6), 131.2 (C5), 128.3/127.3 (Ar–C o/m), 126.9 (Ar–C, p), 69.4 (C3), 59.1 (C8), 57.3 (C1), 51.2 (C12), 40.4 (C7), 39.8 (C4), 23.5 (C9).
| n | M n [kDa] | M w [kDa] | PDI |
|---|---|---|---|
| 100 | 31.5 | 39.8 | 1.26 |
| 150 | 35.2 | 53.7 | 1.52 |
| 200 | 36.0 | 67.1 | 1.86 |
| 300 | 60.3 | 131.2 | 2.17 |
Results and discussion
Monomer synthesis and characterization
The monomer AzaN was synthesized from the hetero Diels–Alder reaction of methylglyoxylate hemiacetal, R-1-methylbenzylamine, and cyclopentadiene in a one-pot synthesis (Scheme 2). A low reaction temperature of −60 °C was maintained throughout the synthesis in order to optimize the yield of the kinetically favoured product. 1H-NMR, 13C-NMR and FT-IR spectra of purified AzaN confirmed its successful synthesis, while optical rotation of [α]20D = +113.2° additionally revealed enantiomerically selective synthesis (Experimental section). Notably, previous literature reports focussed on the monomer with an inverted optical rotation of [α]20D = −112.3°.24 According to Waldmann and Braun, the chemical shift of the proton of the tertiary carbon atom of the azanorbornene ring in the vicinity of the nitrogen atom was lower than 4 ppm.15 NOESY measurements with a selective band center at 2.49 ppm revealed signal enhancements between the proton of the carbon atom bearing the methylcarboxylate group and one olefinic proton as well as between the proton of the tertiary carbon atom of the phenylethyl group and one olefinic proton. 1D-NOESY measurements with a selective band center at 3.54 ppm indicated that the proton of one tertiary carbon atom of the azanorbornene ring itself was located in the vicinity of one olefinic proton, enabling the distinction between the two olefinic protons. Hence, the endo/exo-selectivity of the hetero Diels–Alder reaction could be verified. Due to the symmetry of the C5H6-cycle fragment, however, it could not be reliably determined from NMR and IR analyses whether the nitrogen was located at the 2- or 3- position. In order to unambiguously determine the position of the nitrogen atom in AzaN, single crystals of the compound were examined by single crystal X-ray diffraction. AzaN crystallizes in the monoclinic space group P21 with Z = 2 formula units in the unit cell. The asymmetric unit contains one formula unit (Fig. 1). Differing from previous literature reports, the nitrogen atom was found in position 2 instead of 3, which was attributed to the low temperature maintained during the synthesis and the inherent kinetic control of the monomer synthesis.24 Notably, in the crystalline phase, the R-1-methylbenzyl substituent at the nitrogen atom was found exclusively in the endo position. | ||
| Scheme 2 Reaction scheme for the synthesis of diastereoselectively pure AzaN from methylglyoxylate hemiacetal, R-1-methylbenzylamine, and cyclopentadiene as well as atomic position numbering (in gray) and labelling of chiral C atoms (asterisks). | ||
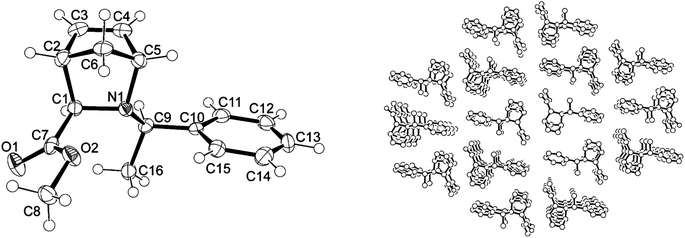 | ||
| Fig. 1 Asymmetric unit in the structure of AzaN (left; ORTEP drawing with 50% probability ellipsoids and atomic numbering). Selected bond lengths (Å) and angles (°): N1–C1 1.487(3), C1–C2 1.562(3), C2–C3 1.514(3), C3–C4 1.321(3), C4–C5 1.518(3), C5–N1 1.507(3), C2–C6 1.539(3), C6–C5 1.519(3); C1–N1–C5 103.75(17), N1–C5–C4 109.45(19), C5–C6–C2 92.13(18). Packing diagram in the crystalline phase of AzaN (right). | ||
The absolute configuration of AzaN (Scheme 2) hence can be explained by (i) the attack of the imine by cyclopentadiene from the Si site (due to the bulky aromatic moiety at the Re site), (ii) the stabilization of the transition state by the alignment of the double bonds of cyclopentadiene with the carbonyl bonds of the methyl acetate group, and (iii) the preference for exo products in asymmetric Diels–Alder reactions, which seems to be favored by the bulky substituent at the nitrogen atom in the endo position, despite the possible inversion at the nitrogen atom.13
Quantum chemical calculations were performed on the monomer itself and the most simple “polymer” model (represented by one repeating unit) using the Gaussian 09 package in order to estimate the stability of the endo position of the substituent at the nitrogen atom (Scheme 3).31 For the monomer, a systematic conformational analysis was carried out using the Austin model 1 (AM1) method.32 The geometries for all conformers were further optimized at the density functional theory (DFT) level choosing the PBE0 functional and the 6-311++G** basis set.33,34 All DFT calculations were performed with acetonitrile as solvent, invoking the polarized continuum model.35 It was found that the monomer has two low-energy conformations, both of them endo isomers, differing in the position of the –COOCH3 group. The remaining conformations were much higher in energy, and consequently not favored at room temperature. The optimized geometry for the most stable conformer strongly resembled the structure determined by X-ray crystal structure analysis (Fig. 1 and 2). Analogously, the geometries for the corresponding conformers of the polymer model were also optimized at the DFT level, including exo–endo combinations of the double bonds in addition to the exo–exo and endo–endo combinations standardly formed in metal-catalyzed ROMP. The results showed that the most stable conformer of the molecule exhibited exo conformation at the nitrogen atom and endo configuration for both double bonds (Fig. 2). The ratio of exo and endo conformers at the nitrogen atom at room temperature was calculated to be roughly 2:1; hence, in the polymer, most repeating units exhibit exo conformation at the nitrogen atom.
 | ||
| Scheme 3 Repeating units of the polymer considered for optimization at the DFT level showing all exo–endo combinations of the olefinic groups. The substituent at the nitrogen atom was considered in exo- as well as in endo position. | ||
 | ||
| Fig. 2 Most stable conformers of the monomer (left) and the repeating unit (right) according to DFT calculations. While in the monomer the substituent at the nitrogen atom shows a preference for the endo position, in the repeating polymer unit it shows a preference for the exo position. | ||
Polymer synthesis and characterization
Diastereomerically pure AzaN could be polymerized with the Ru-based Umicore M31® catalyst at room temperature (Scheme 1): the reactions with targeted polymerization degrees of 100 and 150 yielded pAzaNn with reasonably symmetrical average molecular weight distributions and polydispersity indices of 1.26 and 1.52, respectively. However, the reactions aiming at higher polymerization degrees, namely 200 and 300, required the addition of a second amount of catalyst, concomitant with significant broadening of the average molecular weight distributions (which exhibited a bimodal shape due to re-initiation of the polymerization upon the second catalyst addition), represented by polydispersity indices of 1.86 and 2.17 (Table 1). Nonetheless, it could be verified that the latest 3rd generation ruthenium metathesis initiators can be used to polymerize nitrogen-bearing norbornenes. The stereoregularity of the polymers (Scheme 1) was investigated by 1H-NMR and 13C-NMR spectroscopy in CDCl3 as reported in the literature.36–38 The broad single peak at δ = 5.49 ppm in the one-dimensional 1H-NMR spectrum concomitant with the absence of cross-coupling of the olefinic protons in the 1H-COSY spectrum (Fig. 3) indicated that the olefinic protons were in reciprocally identical environments, which indicated tail–tail connections of adjacent monomer units in the polymer. | ||
| Fig. 3 One-dimensional 1H-NMR spectrum of pAzaN300 with excerpts from the corresponding two-dimensional 1H-NMR COSY spectrum as insets (left). 13C-NMR spectrum of pAzaN300 exhibiting a single set of lines (right). | ||
The cis/trans regiochemistry of the polymers could be determined by detailed 13C-NMR analysis: the 13C-NMR spectrum (Fig. 3) showed a single set of signals representing a highly stereoregular polymer. pAzaNn shows high cis selectivity: in the region of 30–70 ppm, no (residual) signals representing transpAzaNn could be detected. In addition, the infrared spectra of pAzaNn revealed strong absorption in the region 718–676 cm−1 (cis CC bonds) and did not exhibit absorption in the region 980–955 (trans CC bonds). As a final consequence, pAzaNn prepared with the Umicore M31® catalyst has a highly ordered cis-TT syndiotactic microstructure. The Mo-based initiators previously described in the literature yielded isotactic poly(azanorbornene)s (Scheme 1).24–26
Secondary structure determination of the solubilized polymer
Acetonitrile was chosen as the solvent for the induction of secondary (solution) structures due to its aprotic character and its medium polarity. The secondary solution structure of pAzaN300 was determined by circular dichroism (CD) measurements and atomic force microscopy (AFM) and confirmed by theoretical modelling. Vertical excitation energies as well as oscillator and rotator strengths (in the velocity gauge) were calculated for the low-energy conformers of the monomer and the polymer model using the time-dependent DFT method with the same functional and basis set used for the modeling of the monomer and the repeating unit, namely the PBE0 functional and the 6-311++G** basis set.32,33,39The theoretical absorption and CD curves (Fig. 4) were calculated as superpositions of individual Gaussian functions centered at the wavelengths of the theoretically calculated transitions and having heights proportional to the corresponding calculated oscillator and rotator strengths, respectively. The spectra of the individual conformers were Boltzmann-weighted, and the simulated spectra were normalized so that the height of the largest peak superimposed with that of the experimental spectra.
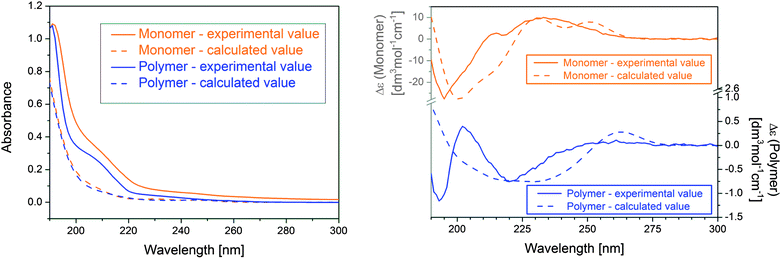 | ||
| Fig. 4 Theoretical and experimental absorption (left) and CD spectra (right) of AzaN and pAzaN300. | ||
The differing CD signals of the monomer and polymer indicated additional chiral elements of the polymer in the solution (Fig. 4). The satisfactory agreement of the experimental and theoretical absorption and CD spectra of the monomer verified the applied theoretical model. The difference between the measured and calculated CD spectra of the simple polymer model suggested that the measured CD spectrum was not simply the superposition of the spectra of individual repeating units: either the chromophores of the repeating units interacted and/or some chiral secondary structure developed in the solution, which was responsible for these additional features in the CD spectrum. AFM images (Fig. 5) additionally backed up the assumption of secondary structure formation in the solution. In comparison with blank samples of spincoated acetonitrile, the analogously spincoated solution of pAzaN300 (both on silicon wafers) exhibited unbent polymer rods of very similar dimensions in the range of 100 × 30 × 2.5 nm that were arranged in reciprocally parallel alignment, most likely due to the spincoating of the polymer solution. (“One” rod had a dimension of 210 × 30 × 2.5 nm and was seemingly composed of two individual rods.) Within the resolution of the AFM tip of approx. 10 nm, no deviation from a straight alignment could be detected. These results clearly evidenced the formation of regular secondary polymer structures in acetonitrile solution.40 The dimensions of the rods significantly deviated from cylindrical shapes with circular area and moreover exhibited large diameters of an ellipsoid/circular basic projection structure.
 | ||
| Fig. 5 AFM topography (left) and phase (right) images of a spincoated solution of acetonitrile (top) and a spincoated solution of pAzaN300 in acetonitrile (bottom). The polymer formed unbent rods of the approximate dimensions 100 × 30 × 2.5 nm (circumframed by ellipsoids). The shape at the bottom of the image seemingly is composed of two individual rods. | ||
In order to gain some insight into the possible secondary structures of the polymer, a polymer fragment of eight repeating units (using the most stable conformer of the simple polymer model) was constructed. The monomer units were arranged in a cis-TT syndiotactic fashion according to the NMR experiments; the geometry of the model system was optimized by the AM1 method (Fig. 6). The presence of a regular secondary structure was supported by this eight-monomer polymer model, in which the monomers, connected by rigid double bonds with distances of 5.3 Å, formed a right-handed curvature. While the eight-monomer polymer model did not provide enough data for the determination of the gang height and diameter(s) of the secondary structure, it nonetheless allowed estimating some basic geometry of the secondary structure: Within height changes of 0.2 nm only (z axis), the centers of the eight monomers spanned an area of x/y = 3/0.8 nm (Fig. 6), confirming the large diameters observed in the AFM measurements. These large diameters, at least in part, originated from the rigidity of the cis-TT main chain.
 | ||
| Fig. 6 Projection along the calculated secondary structure formed of eight repeating units. For a more lucid presentation, the atoms of the main-chain are represented by the ball-and-stick model, the substituents are displayed in the all-stick model (left). Coordinates of the “center points” of the eight monomers; the center points were calculated as the arithmetic values of the centers of the double bonds (right; all axes are drawn reciprocally proportional). | ||
It must be assumed that (partial) deformations of the solution structure originated from the centripetal forces and solvent evaporation during the spincoating procedure as well as from the AFM tip during the measurement itself. The deformations of the solution structure have been described in detail for paired helical filaments.40 According to CD and AFM measurements, a secondary solution structure of the polymer was formed. The huge diameters observed would be unstable for a single-strand helix of any circular/ellipsoid projection symmetry in a one-solvent solution due to the lack of interhelical stabilization. Hence, a more complex (multiple-strand) solution structure must be considered.
Conclusions and outlook
Successful employment of a ruthenium-based catalyst, namely M31®, in the ROM polymerization of 2-azanorbornenes could be demonstrated for the first time. Monomodal molecular weight distributions of the poly(2-azanorbornene)s, concomitant with polydispersity indices of 1.26 and 1.52, were indicative of the control of the polymerization for PD = 100 and 150; polymerizations aiming at degrees of 200 and 300, on the other hand, required the addition of a second portion of catalyst, concomitant with a broadening of the molecular weight distribution in bimodal shape. The poly(2-azanorbornene)s prepared using the M31® catalyst had a structure with a high degree of cis-TT stereoregularity, completing the list of four possible regular structures for poly(2-azanorbornenes) by a third entry in addition to the cis-HT and trans-HT stereoregular structures obtained from molybdenum-catalyzed ROM polymerizations. The formation of a regular secondary structure in acetonitrile solution could be shown by CD and AFM measurements (the latter of spincast polymer samples) and was also supported by AM1 calculations of a polymer model of eight repeating units. The precise nature of the helical structure and its use in molecular recognition strategies will be a subject of further investigations involving 2-azanorbornene monomers with different substitution patterns.Acknowledgements
This study was performed at the Institute for Chemistry and Technology of Materials (ICTM) of the Graz University of Technology with funding and contributions from the Polymer Competence Center Leoben GmbH (PCCL, Austria) within the framework of the COMET program of the Austrian Ministry of Traffic, Innovation and the Ministry of Economy, Family and Youth, acknowledged by F.S. and F.W. PCCL is funded by the Austrian Government and the State Governments of Styria and Upper Austria. M.K. acknowledges the financial support provided by the European Research Council (ERC) under the European Community's Seventh Framework Programme (FP7/2007-2013), ERC Grant Agreement no. 200639, and the Hungarian Scientific Research Fund (OTKA), grant no. NF72194. Karl Rametsteiner of KEKELIT GmbH is gratefully acknowledged for the provision of methylglyoxylate hemiacetal.Notes and references
- M. R. Buchmeiser, Chem. Rev., 2000, 100, 1565–1604 CrossRef CAS.
- Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vol. 1–3 Search PubMed.
- R. R. Schrock, Chem. Rev., 2002, 102, 145–180 CrossRef CAS.
- T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS.
- G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS.
- M. M. Flook, A. J. Jiang, R. R. Schrock, P. Müller and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 7962–7963 CrossRef CAS.
- M. Lichtenheldt, D. Wang, K. Vehlow, I. Reinhardt, C. Kühnel, U. Decker, S. Blechert and M. R. Buchmeiser, Chem.–Eur. J., 2009, 15, 9451–9457 CrossRef CAS.
- W. H. Binder, S. Kurzhals, B. Pulamagatta, U. Decker, G. M. Pawar, D. Wang, C. Kühnel and M. R. Buchmeiser, Macromolecules, 2008, 41, 8405–8412 CrossRef CAS.
- H. Sunden, I. Ibrahem, L. Eriksson and A. Cordova, Angew. Chem., Int. Ed., 2005, 44, 4877–4880 CrossRef CAS.
- A. A. Ruggiu, R. Lysek, E. Moreno-Clavijo, A. J. Moreno-Vargas, I. Robina and P. Vogel, Tetrahedron, 2010, 66, 7309–7315 CrossRef CAS.
- O. Diels and K. Alder, Justus Liebigs Ann. Chem., 1928, 460, 98–122 CrossRef CAS.
- P. D. Bailey, R. D. Wilson and G. R. Brown, J. Chem. Soc., Perkin Trans. 1, 1991, 1337–1340 RSC.
- L. Stella, H. Abraham, J. Feneau-Dupont, B. Tinant and J. P. Declercq, Tetrahedron Lett., 1990, 31, 2603–2606 CrossRef CAS.
- H. Abraham and L. Stella, Tetrahedron, 1992, 48, 9707–9718 CrossRef CAS.
- H. Waldmann and M. Braun, Liebigs Ann. Chem., 1991, 1045–1048 CrossRef CAS.
- A. Leitgeb, J. Wappel and C. Slugovc, Polymer, 2010, 51, 2927–2946 CrossRef CAS.
- N. Watkins, P. Quigley and M. Orton, Macromol. Chem. Phys., 1994, 195, 1147–1164 CrossRef CAS.
- K. Melis, D. De Vos, P. Jacobs and F. Verpoort, J. Mol. Catal. A: Chem., 2011, 169, 47–56 CrossRef.
- K. J. Ivin, L. M. Lam and J. J. Rooney, Makromol. Chem., 1993, 194, 3203–3207 CrossRef CAS.
- C. Larroche, J. P. Laval, A. Lattes, M. Leconte, F. Quignard and J. M. Basset, J. Org. Chem., 1982, 47, 2019–2026 CrossRef CAS.
- G. W. Coates and R. M. Waymouth, J. Mol. Catal., 1992, 76, 189–194 CrossRef CAS.
- R. R. Schrock, Dalton Trans., 2011, 40, 7484–7495 RSC.
- M. M. Flook, V. W. L. Ng and R. R. Schrock, J. Am. Chem. Soc., 2011, 133, 1784–1786 CrossRef CAS.
- R. M. E. Schitter, T. Steinhäusler and F. Stelzer, J. Mol. Catal. A: Chem., 1997, 115, 11–20 CrossRef CAS.
- J. G. Hamilton, K. J. Ivin and J. J. Rooney, J. Mol. Catal. A: Chem., 1986, 36, 115–125 CAS.
- F. Stelzer, R. M. E. Schitter and T. Steinhäusler, NATO ASI Ser., Ser. 1, 1995, 506, 243–252 Search PubMed.
- P. Preishuber-Pflügl, P. Buchacher, E. Eder, R. M. E. Schitter and F. Stelzer, J. Mol. Catal. A: Chem., 1998, 133, 151–158 CrossRef.
- G. M. Sheldrick, SHELXS-97, Program for structure solution, University of Göttingen, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for crystal structure analysis, University of Göttingen, 1997 Search PubMed.
- D. Burtscher, C. Lexer, K. Mereiter, R. Winde, R. Karch and C. Slugovc, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4630–4635 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. CrossV. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09, Revision B.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- M. J. S. Dewar, E. G. Zoebisch and E. F. Healy, J. Am. Chem. Soc., 1985, 107, 3902–3909 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS Erratum, J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef.
- C. Adamo and V. J. Barone, Chem. Phys., 1999, 110, 6158–6170 CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- J. G. Hamilton, K. J. Ivin and J. J. Rooney, Br. Polym. J., 1984, 16, 21–33 CrossRef CAS.
- K. M. Totland, T. J. Boyd, G. G. Lavoie, W. M. Davis and R. R. Schrock, Macromolecules, 1996, 29, 6114–6125 CrossRef CAS.
- R. R. Schrock, Acc. Chem. Res., 1990, 23, 158–165 CrossRef CAS.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS.
- F. Moreno-Herrero, M. Perez, A. M. Baro and J. Avila, Biophys. J., 2004, 86, 517–525 CrossRef CAS.
Footnote |
| † CCDC [876454]. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c2py20219k |
| This journal is © The Royal Society of Chemistry 2012 |