Copper mediated controlled radical polymerization of methyl acrylate in the presence of ascorbic acid in a continuous tubular reactor
Nicky
Chan
,
Michael F.
Cunningham
* and
Robin A.
Hutchinson
*
Department of Chemical Engineering, Queen's University, Kingston, ON, Canada K7L 3N6. E-mail: michael.cunningham@chee.queensu.ca; robin.hutchinson@chee.queensu.ca
First published on 23rd March 2012
Abstract
Controlled radical polymerization of methyl acrylate catalyzed by copper was conducted in a continuous tubular reactor. A length of copper tubing was used to initiate polymerization and generate soluble copper species, while the bulk of polymerization took place in inert stainless steel tubing. To mediate polymerization in the absence of copper surface, environmentally benign ascorbic acid was used for the first time in single electron transfer-living radical polymerization (SET-LRP) as a reducing agent to regenerate activating copper species. Polymerizations were conducted at ambient temperature with 30 wt% DMSO as solvent, producing well defined living polymer at a steady state conversion of 78% for a residence time of 62 min. Chain extensions using outlet polymer solutions were well-controlled and proceeded to high conversion in a short period of time, with a final concentration of 10 ppm of residual copper. The results illustrate the significant potential of using a continuous tubular reactor with ascorbic acid as a reducing agent as an efficient means to scale-up production of well controlled polyacrylics and other multiblock copolymers.
Introduction
Controlled radical polymerization (CRP) has been the subject of intense academic interest as an attractive route towards the synthesis of novel polymeric materials that cannot be produced using conventional free radical polymerization methods. Atom transfer radical polymerization (ATRP) is an effective and dynamic technique for the controlled polymerization of many vinyl monomers under mild conditions.1–4 A transition metal catalyst is used in ATRP to mediate reversible activation and deactivation of dormant and growing radicals. Conventional ATRP utilises copper(I) and copper(II) salts with nitrogen based ligands in stoichiometric or slight sub-stoichiometric ratios relative to the initiating species, which results in a high catalyst concentration on the order of 104 parts per million (ppm) in the final polymer. This high concentration of transition metal leads to difficulties in process design as post polymerization treatments are required to remove residual catalyst which is often toxic and adds undesired colour to the final product.5 This complication imposes increased production cost, and has hindered the adoption of ATRP on an industrial scale.6–9Improved understanding of the electrochemistry involved in ATRP has led to the development of more active catalyst complexes and novel ATRP techniques such as “activator regenerated by electron transfer” (ARGET),10 “initiators for continuous activator regeneration” (ICAR),11 and “single electron transfer-living radical polymerization” (SET-LRP).12,13 All three variants allow for reduction in catalyst concentrations by 2–3 orders of magnitude to below 102 ppm levels in the final polymer. Of the three techniques, ARGET and ICAR are most similar to conventional ATRP, with a reducing agent or a free radical initiator added to the polymerization to regenerate copper(I) activating species from copper(II) deactivators that have accumulated due to bimolecular radical termination.
At first glance, SET-LRP bears a strong resemblance to ATRP, as copper species are used in both systems to govern the equilibrium between activation and deactivation species. However, Percec et al. propose that in SET-LRP, copper(0) acts an activating species via outer sphere electron transfer to form transient copper(I) species.14 Under appropriate combinations of polar or coordinating solvents (DMSO, alcohols, water) and ligands that favour disproportionation, copper(I) species undergo near instantaneous disproportionation into copper(0) and copper(II) species to form activators and deactivators in pairs.15,16 This catalytic cycle is proposed to give greater control over the polymerization and retention of chain end fidelity compared to ATRP, for which copper(I) species are the primary activators via inner sphere electron transfer.17
Recent studies into SET-LRP have suggested that copper(0) has a dual role as reducing agent and activator.18–20 The duality of copper(0) suggests that it may be possible to utilise reducing agents common to ARGET ATRP to enhance the polymerization rate and increase tolerance to air in SET-LRP by changing the amount of reducing agent that is added. It has been established that changing the ratio of reducing agent to copper species can alter the ratio of activating to deactivating species, to increase the rate of polymerization in ATRP.21–24 There has been very little research done, however, combining SET-LRP with reducing agents. It was found that SET-LRP polymerizations with hydrazine, a commonly used reducing agent in ATRP, showed a higher rate of polymerization as well as increased tolerance to air.25,26 However, the mode of activation for hydrazine was attributed to reduction of Cu2O generated by the oxidation of copper(0) with air in the system. The use of hydrazine was later extended to activation of copper(0) surface to increase the rate of polymerization.27 Unfortunately, hydrazine is a highly reactive and toxic chemical and poses a hazard even in small quantities. In a previous study, we were able to re-initiate a SET-LRP reaction using a non-toxic reducing agent, tin(II) 2-ethylhexanoate, in the absence of additional copper(0) metal.28 While the effect of reducing agent concentration was not investigated, the results demonstrate the potential for other non-toxic and environmentally benign reducing agents such as ascorbic acid to be beneficial for SET-LRP.
Although the mechanistic nature of SET-LRP is still a subject of scrutiny in literature,12,14,15,29,30 it has been established that SET-LRP processes offers several advantages when compared with ATRP systems. Compatibility with polar solvents such as alcohols and water offer the use of more environmentally friendly reaction media, in addition to making SET-LRP more suitable for the polymerization of water-soluble or amphiphilic materials. Catalyst can be added in the form of elemental copper powder or wire, greatly simplifying catalyst handling, removal and recycling.15,31 Furthermore, it can polymerize acrylics at low temperature with excellent control and much faster reaction rates than observed in ATRP. Soeriyadi et al. have used this level of enhanced control and reactivity to synthesize high-order multi-block copolymers via an iterative SET-LRP procedure.32 The polymerizations proceeded to high conversion, and it was possible to polymerize additional blocks without isolating the existing polymer, greatly simplifying the number of steps required when compared to ATRP or other CRP techniques.
At the same time, SET-LRP is not without its own set of issues. Induction times in laboratory scale reactions are occasionally observed which can lead to batch to batch variability in polymer properties under the same process parameters.31,33 Additionally, due to the large heat release associated with rapid polymerization, SET-LRP reactions are highly exothermic. Levere et al. observed internal temperatures in excess of 60–100 °C for SET-LRP of methyl acrylate conducted initially at room temperature in a small Schlenk tube (working volume of approximately 25 mL) due to insufficient heat removal.33 This behaviour poses risk of runaway reaction when polymerization is scaled to larger volumes, and might make it difficult to conduct SET-LRP in batch reactors on a commercial scale.
While there is only a limited number of publications studying the use of ATRP in continuous processes,24,34–38 the engineering challenges surrounding SET-LRP can be readily remedied by the use of continuous reactors. Inconsistencies between start-up are conveniently eliminated in continuous processes, and once at steady state, continuous processes yield more consistent product quality and higher productivity than comparable batch reactors. Continuous flow tubular reactors in particular are ideal for scale up due to their large surface area to volume ratio, which greatly improves heat removal and temperature control. We have recently demonstrated the use of copper tubing as both reactor and catalyst source in the design of a continuous tubular reactor for SET-LRP.28 Although the proof of concept was a success, some concerns remained about the long term structural integrity of the reactor, as copper was leached from the reactor walls into solution. In addition, to synthesize multi-block copolymers in a continuous process, a higher conversion would need to be reached in a reasonable residence time.
In this work, we aim to improve upon the original reactor design and address some of the shortcomings observed. To that end, a combination of copper tubing and stainless steel tubing is used to construct a continuous tubular reactor. A short copper coil is used to initiate polymerization and generate soluble copper species in situ. The majority of the reaction is then carried out in a longer segment of inert stainless steel tubing. The effects of environmentally benign and non-toxic ascorbic acid to enhance the rate of SET-LRP in the absence of copper(0) surface while maintaining good control over chain end fidelity are investigated in batch and continuous reactors and reported for the first time. The combination of an environmentally friendly additive that can be used in combination with “green” solvents, along with an improved and more robust process design makes a continuous tubular reactor an ideal platform for the synthesis of novel polymeric materials.
Experimental
Materials
Methyl acrylate (MA) (99%, inhibited by less than 100 ppm monomethyl ether hydroquinone, Aldrich), dimethyl sulfoxide (DMSO) (99.9%, Aldrich), methyl 2-bromopropionate (MBP) (98%, Aldrich), L-ascorbic acid (AscA) (99%, Aldrich), and nitrogen gas (Praxair, 99.998%) were used as received. Tris[2-(dimethylamino)ethyl]amine (Me6TREN) was synthesized as described previously.39Continuous tubular reactor setup
A schematic of the reactor setup is shown in Fig. 1. The reactor was split into two sections. Section 1 was composed of an Eldex Recipro VS series high pressure liquid metering pump (B-125-VS), capable of feeding between 0.2 to 10.0 mL min−1. The pump was connected to a series of refrigeration grade copper tubing (3.2 mm outer diameter, 1.65 mm inner diameter). The tubing was split into four pieces of equal length, for a total length of 15.24 m and a total volume of approximately 32 mL. The modular nature of the copper tubing allowed for facile assembly into reactor configurations ranging from 8 to 32 mL. A 500 mL three neck round bottom flask was used as a feed vessel, and the feed solution was kept stirring under nitrogen.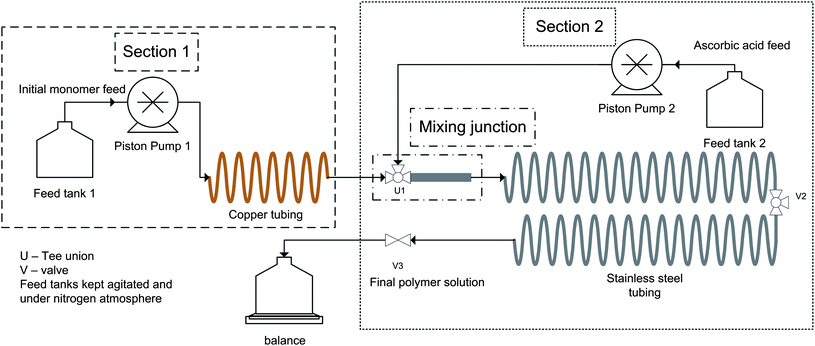 | ||
| Fig. 1 Schematic of continuous tubular reactor used for copper(0) mediated polymerization. | ||
The outlet solution from the copper tubing was mixed continuously with material from Feed Tank 2 in a three way Tee union U1, designated as the beginning of Section 2. The flow rate of the second feed was also controlled by an Eldex Recipro VS series high pressure liquid metering pump (B-125-VS). U1 was connected to a 10 cm section of stainless steel tubing (6.35 mm outer diameter, 4.57 mm inner diameter). A small piece of copper wire (0.155 mm in diameter, 1.44 g) was inserted into the tubing and valve to act as a baffle and enhance mixing between the two feeds. This section of the reactor was designated as the mixing junction as shown in the schematic. A narrower set of stainless steel tubing (15.24 m in length, 3.2 mm outer diameter, 2.1 mm inner diameter) was connected to the mixing junction. The total mass flow rate out of the stainless steel tubing was controlled as the sum of the two metering pumps, and the flow rate was measured using a Mettler Toledo PG 5002s balance. The entire reactor configuration was kept in a fume hood and all polymerizations were done at ambient temperature (23–25 °C). The copper coils and copper wire baffle were weighed before and after each experiment, and mass loss was determined to be negligible (<0.1% of total weight).
Synthesis procedure for continuous SET-LRP polymerization in copper tubing
MA (210 g, 2.44 mol), DMSO (90 g, 1.15 mol), Me6TREN (0.046 g, 0.244 mmol), and MBP (4.08 g, 24.4 mmol) were combined in a 500 mL three neck round bottom flask for a ratio of reactants [MA]0:[MBP]0:[Me6TREN]0 of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.01. The initial recipe gave a target degree of polymerization of 100, a target number-average molecular weight (Mn) of 8640 g mol−1 and a polymer content of 70 wt% at full conversion. The feed solution was kept agitated and purged with nitrogen for approximately 1 h before the start of polymerization. At the same time, the copper tubing was flushed with nitrogen to minimize inhibition from the presence of oxygen in the system.
:1:0.01. The initial recipe gave a target degree of polymerization of 100, a target number-average molecular weight (Mn) of 8640 g mol−1 and a polymer content of 70 wt% at full conversion. The feed solution was kept agitated and purged with nitrogen for approximately 1 h before the start of polymerization. At the same time, the copper tubing was flushed with nitrogen to minimize inhibition from the presence of oxygen in the system.
After deoxygenation, the reactants were pumped into the copper tubing at a high flow rate to fill the reactor, typically taking 2–4 min. Once the reactor was filled, the flow rate was reduced to 1 g min−1. This action was considered the start of the polymerization. The flow rate was carefully monitored over the course of the polymerization, and samples were taken at specified time intervals from the outlet of the copper tubing (outlet of Section 1).
Synthesis procedure for continuous SET-LRP with entire reactor
As with the procedure above, a feed solution was prepared for Section 1 of the reactor. MA (175 g, 2.03 mol), DMSO (75 g, 0.96 mol), Me6TREN (0.038 g, 0.203 mmol), and MBP (3.40 g, 20.3 mmol) were combined in a 500 mL three neck round bottom flask. The feed solution (F1) was stirred and purged with nitrogen for approximately 1 h before the start of polymerization. At the same time, MA (175 g, 2.03 mol), DMSO (75 g, 0.96 mol), and ascorbic acid (0.036 g, 0.203 mmol) were combined in another 500 mL three neck round bottom flask. This second feed solution (F2) was also stirred and purged with nitrogen for approximately 1 h before the start of polymerization.While the feed solutions were being purged, nitrogen was flushed through both the copper and stainless steel tubing to minimize the impact of oxygen in the system. After the feed solutions were deoxygenated, the first feed solution F1 containing monomer, ligand and initiator was fed into the copper tubing at a flow rate of 1 g min−1. After approximately 15 min, the copper tubing, the mixing junction, and the inlet section of the stainless steel tubing have all been saturated with reagents. At that time, feeding of F2 was started at the same flow rate of 1 g min−1. This action was considered the start of the polymerization. The total flow rate out of the reactor was carefully monitored to be the sum of the individual metering pumps for a total of 2 g min−1. Samples were taken at specified time intervals from the outlet of the reactor. The target molecular weight of the polymerization was 17280 g mol−1 (DPn = 200), for a ratio of reactants [MA]0:[MBP]0:[Me6TREN]0:[AscA]0 of 200:1:0.01:0.01. Solvent content was 30% by mass.
SET-LRP batch polymerization procedure with ascorbic acid
A few batch polymerizations were conducted to study the effects of adding ascorbic acid to a SET-LRP reaction. MA (35 g, 0.407 mol), DMSO (12.5 g, 0.16 mol), Me6TREN (0.0077 g, 0.0407 mmol), and MBP (0.68 g, 4.07 mmol) were combined in a 100 mL three neck round bottom flask. The mixture was purged under nitrogen for one hour, before being submerged in an oil bath set to 30 °C. Ascorbic acid (7.2 mg, 0.0407 mmol) was dissolved in 2.5 g of DMSO, purged with nitrogen for ten minutes and then transferred to the round bottom flask via a degassed syringe. A piece of copper wire (0.155 m in diameter, 2.024 g) was gently scrubbed with acetone and a non-abrasive material to remove any surface residue, wrapped around a stir bar and then added to the flask to initiate polymerization. Samples were withdrawn using a deoxygenated syringe at specified time intervals, cooled and exposed to air to stop polymerization. The target molecular weight of the polymerization was 8640 g mol−1 (DPn = 100). Solvent content was 30% by mass.Synthesis procedure for chain extension of outlet polymer solution
Chain extensions using outlet solution from the tubular reactor were conducted to verify the livingness of the polymer produced. 25 g of polymer solution from the outlet was added to a 100 mL three neck round bottom flask. MA (17.5 g, 0.203 mol) and DMSO (5 g, 0.064 mol) were also added to the flask. The polymer solution was then purged with nitrogen for 1 h and placed into an oil bath set at 30 °C. Me6TREN (7.7 mg, 0.0407 mmol) was mixed with 1 g of DMSO, purged with nitrogen and then injected into the system via a degassed syringe. After approximately 10 min, ascorbic acid (1.8 mg, 0.010 mmol) dissolved in 1.5 g of DMSO purged with nitrogen was also injected via a degassed syringe to reinitiate polymerization. Samples were withdrawn using a deoxygenated syringe at specified time intervals.Analytical methods and characterization
Sample conversion was determined by gravimetry. Gel permeation chromatography (GPC) was used to determine the molecular weight distribution of the polymer samples. Samples were prepared by dissolving 30 mg of dried polymer in 3 mL of THF. The dissolved samples were then passed through a column packed with basic alumina to remove any remaining copper, before being filtered through a nylon filter (0.2 μm pore size). The GPC was equipped with a Waters 2960 separation module containing four Styragel columns of pore sizes 100, 500, 103, 104 Å, coupled with a Waters 410 differential refractive index (RI) detector (930 nm) operating at 40 °C. THF was used as eluant and the flow rate was set to 1.0 mL min−1. The detector was calibrated with eight narrow polystyrene standards ranging from 347 to 355 000 g mol−1. The molecular weights of poly(MA) samples were obtained by universal calibration with Mark-Houwink parameters for polystyrene (K = 11.4 × 10−5 dL g−1, a = 0.716) and poly(MA) (K = 6.11 × 10−5 dL g−1, a = 0.799) at low molecular weights.40Residual copper concentration measurements
Residual copper concentration in the polymer solution was measured by flame atomic absorption spectroscopy using a Varian Spectra AA-20 Plus Flame Atomic Absorption Spectrometer, with a copper lamp set to a wavelength of 324.9 nm. The outlet polymer solution was diluted in methyl isobutyl ketone (MIBK) at a ratio of 1:20. Calibration standards were prepared from 500 ppm 21 Elemental Standard in MIBK in a range of dilutions from 0.5 ppm to 5.0 ppm. A calibration check solution was prepared from 300 ppm 21 Element Standard at a concentration of 3.0 ppm.
Results and discussion
Limitations of an all copper tubular reactor
As briefly discussed in the introduction, there were three impediments for use of the original all copper tubular reactor for large scale production of polymeric materials:1. Structural integrity of the reactor as copper leaches from the reactor walls.
2. Reaching a high steady state conversion with a reasonable residence time for the production of block copolymers.
3. Broadening of the molecular weight distribution with operation time due to concentration gradients of reactants across the diameter of the tube at higher conversions.
In the original study,28 the three problems were correlated and difficult to separate. The copper reactor wall acts as a catalyst source for the activation of dormant chains, and activation of a polymer chain or initiating alkyl halide species generates a copper(I) species on the reactor wall which becomes more readily soluble in solution. As such, the amount of copper that leaches into solution is proportional to the number of activations along the length of the reactor. It is possible to limit the amount of soluble copper species by decreasing the ligand concentration, but doing so will generally slow the reaction rate.41
To reach a higher steady state conversion in a continuous tubular reactor, process operating parameters such as temperature or residence time can be altered. Increasing the residence time, either by lowering the flow rate or increasing the length of the reactor, leads to higher conversion.28 However, solution viscosity also increases, slowing transport of copper species across the radius of the reactor. This makes it likely that chains which are closer to the reactor wall and catalyst source will grow more quickly, leading to a population of higher molecular weight chains and broadening the outlet molecular weight distribution, as was observed experimentally.
In addition, increasing the length of copper tubing increases the available surface area for catalysis. As activation can occur anywhere along the reactor wall, the increased length of copper tubing makes it more difficult to detect where structural integrity may be compromised due to copper leaching. Therefore, it is beneficial that the copper surface area and amount of copper which can leach into solution be limited to improve reactor operation and reduce maintenance.
To illustrate this interdependent behaviour, three experiments were conducted in copper tubing of various lengths. The experimental conditions and steady state properties of the poly(methyl acrylate) (pMA) produced are summarized in Table 1. The experiments were conducted with a low ligand ratio of 1:100 with respect to initiator, to limit the amount of soluble copper in order to prolong the longevity of the reactor as well as to reduce the need for removal of residual copper in a post polymerization treatment.
:1:0.01, and 30 wt% DMSO as solvent
| Exp. | Reactor length (m) | Reactor volume (mL) | τ (min) | conva (%) | Mn,theob (g mol−1) | Mn,GPC (g mol−1) | PDI | Ieffc(%) |
|---|---|---|---|---|---|---|---|---|
| a Conversion and molecular weight data taken as average steady state values (±standard deviation) after 3 residence times. b Mn,theo = ([M]0/[MBP]0) × conv. × MWM. c Ieff = Mn,theo/Mn,GPC × 100% as calculated from steady state averages. d Experiment T4 was conducted using a combination of copper tubing (3.81 m) and stainless steel tubing (15.24 m). | ||||||||
| T1 | 3.81 | 8 | 8 | 41 ± 1.4 | 3550 | 3960 ± 150 | 1.48 ± 0.02 | 89 |
| T2 | 7.62 | 16 | 16 | 53 ± 2.0 | 4610 | 5120 ± 370 | 1.37 ± 0.04 | 90 |
| T3 | 15.24 | 32 | 32 | 59 ± 2.5 | 5100 | 5360 ± 180 | 1.45 ± 0.07 | 95 |
| T4d | 19.05 | 62 | 62 | 55 ± 1.8 | 4780 | 5020 ± 160 | 1.34 ± 0.02 | 95 |
In all three experiments, living polymer was produced with high initiator efficiency (89 to 95%), although the molecular weight distributions were broadened due to the relatively low conversion. The changes in conversion, number average molecular weight (Mn), and polymer dispersity index (PDI) as a function of dimensionless residence time (t/τ) are shown in Fig. 2. The three experiments all showed some transient behaviour at the beginning of the experiment, with the reactor reaching steady state after an activation time of approximately 30 min. This type of activation behaviour has also been observed in batch reactions,31,33 and demonstrates the benefit of conducting SET-LRP in a continuous process as opposed to a batch reactor.
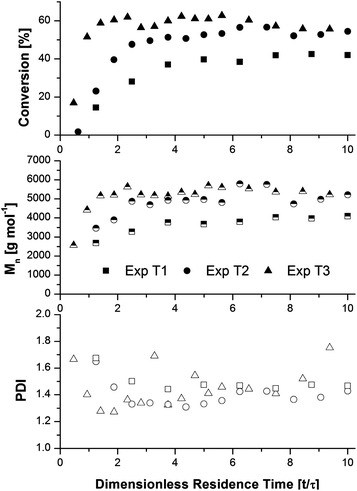 | ||
| Fig. 2 Evolution of conversion (closed symbols), number-average molecular weight (half-filled symbols), and polydispersity index (open symbols) as a function of dimensionless residence time for methyl acrylate SET-LRP polymerizations at ambient temperature in a copper tubular reactor. Experimental conditions are summarized in Table 1. | ||
The data showed that increasing the reactor length and the residence time (τ) from 8 (T1) to 16 (T2) to 32 (T3) minutes gave a corresponding increase in steady state conversion of 41, 53, and 59%, respectively. With increasing conversion an increase in molecular weight was observed, with average steady state Mn values of 3960, 5120, and 5360 g mol−1 for experiments T1–T3. It would seem that a residence time of 16 min (T2) is more favourable for this polymerization recipe, as increasing the residence time to 32 min yields only a small increase in steady state conversion, with the molecular weight distribution becoming broader most likely due to the previously discussed concentration gradients across the radial direction of the reactor.
Extending the reactor using inert stainless steel tubing
A possible means to achieve higher steady state conversion at ambient temperatures with a continuous flow reactor is to use a combination of copper and stainless steel tubing. With this design, a short coil of copper tubing initiates polymerization and generates soluble or “nascent” copper species in situ. The soluble copper species can then mediate the polymerization in a section of the reactor constructed of inert stainless steel. As there is no reaction between the steel walls and polymer chains, the presence of reaction gradients across the radial direction of the reactor are largely eliminated. This concept was demonstrated in experiment T4 by using a short segment of copper tubing (3.81 m) at the beginning of the reactor connected to a longer segment of stainless steel tubing (15.24 m) of the same diameter. Conversion, Mn, and PDI as a function of reaction time for experiment T4 and T1 are compared in Fig. 3. | ||
| Fig. 3 Evolution of conversion (closed symbols), number-average molecular weight (half-filled symbols), and polydispersity index (open symbols) as a function of reaction time for methyl acrylate SET-LRP polymerizations at ambient temperature in a copper and stainless steel tubular reactor. Experimental conditions are summarized in Table 1. | ||
As the residence time of T4 was much longer than experiment T1, it took a longer period of time (>180 min) before the reactor exhibited steady state behaviour. By using an inert material to increase the reactor length, the steady state conversion was increased from 41% (T1) to 55% (T4), with a corresponding increase in Mn from 3960 to 5020 g mol−1. The PDI decreased from 1.48 to 1.34, demonstrating that the increased reactor length did not negatively impact the PDI as observed in experiment T3. This result demonstrates that copper(0), copper(I), and copper(II) species generated in situ are capable of mediating the activation and deactivation equilibrium in the absence of a heterogeneous copper surface.
Interestingly, Lligadas et al. conducted a similar type of experiment in batch where they attempted to resolve the role of nascent copper(0) and copper(I) species, but achieved very different results. In their experiment, two Schlenk tubes under inert atmosphere were connected. A polymerization catalyzed by copper powder was initiated in one tube, and upon reaching 20% conversion the polymer solution was decanted to the second vessel while the copper powder was left in the first flask. In the absence of copper powder, polymerization did not continue within the bounds of experimental error. When the reaction mixture was transferred back to the flask with copper powder, polymerization restarted and reached high conversion in a short period of time. It was concluded that low levels of copper(I) species generated from activation of polymer chains by copper(0) was unable to contribute to chain activation, and that polymerization can only proceed in the presence of an elemental copper surface as an activating species.15
Although contradicting the findings by Lligadas et al., the result from experiment T4 provides some insight into the SET-LRP mechanism. While not possible to identify the primary activating species in the stainless steel section of the reactor, the results suggested that copper(0) was playing a role as both activator and reducing agent as suggested in literature.18,19,29 As ATRP studies have established, bimolecular termination will lead to a build-up of copper(II) deactivating species when a low concentration of copper is used. This accumulation will alter the copper(I) to copper(II) ratio and slow or eventually stop the polymerization. It is likely that the same phenomenon was occurring in the stainless steel tubing. Independent of whether copper(0) or copper(I) was the activating species, a small amount of termination would lead to an increase in copper(II) concentration. Without the presence of a copper surface to act as activator or reducing agent, the equilibrium would thus shift towards deactivation and the build-up of dormant chains. In our previous study on copper(0) mediated CRP, several chain extensions of outlet polymer solution containing in situ generated copper species were conducted.28 The addition of copper wire led to fast activation of dormant polymer chains and polymerization reached high conversion in a short period of time. When no copper wire was added, polymerization did not re-initiate. Most interestingly, the addition of tin(II) 2-ethylhexanoate, a common reducing agent used in ARGET ATRP, was successful in re-initiating the polymerization although the rate of polymerization was slower. Based on this result, it was decided to investigate the effects of ascorbic acid, a stronger, non-toxic and environmentally benign reducing agent in this study.
Effects of ascorbic acid on SET-LRP in batch
Ascorbic acid has been used extensively as a reducing agent in ATRP for the polymerization of many water soluble polymers and in dispersed phase systems.21,42–44 As a reducing agent for ARGET ATRP, it was found that the solvent played a large role in the reaction kinetics and control over polymerization. When ascorbic acid was added in 10–100 fold excess to copper(II) bromide in anisole, the reducing agent remained insoluble and well controlled polymerizations were achieved. However, when a polar solvent like DMF was used, ascorbic acid was fully dissolved and fast reduction of copper(II) deactivating species led to a loss of control over the polymerization.45 Paterson et al. found that for the polymerization of 2-hydroxyethyl methacrylate in methanol, an 80 fold excess of ascorbic to copper(II) bromide was required to achieve a well-controlled polymerization. A 60 fold excess gave only low conversion, and a 120 fold excess led to a loss of control over the reaction.46 As such, it seems that for the polar solvents used in SET-LRP where ascorbic acid will be readily dissolved, a fine balancing act is required to achieve a concentration of ascorbic acid that will yield the desired polymerization kinetics.The experimental conditions and final properties of poly(methyl acrylate) produced by SET-LRP with ascorbic acid are summarized in Table 2. Batch experiments B1 to B3 were conducted with a short length of copper wire as catalyst source. Experiment B1 can be considered the standard batch SET-LRP experiment, showing good control over the molecular weight distribution. The experiment reached a conversion of 73% after 180 min, with a final molecular weight of 8140 g mol−1 and a PDI of 1.28. As the available copper surface area was much lower in a batch vessel, the rate of polymerization in experiment B1 was lower than a comparable reaction in a tubular reactor.
| exp. | [M]0:[MBP]0:[Me6TREN]0:[AscA]0 | t (min) | conv (%) | Mn,theoa (g mol−1) | Mn,GPC (g mol−1) | PDI |
|---|---|---|---|---|---|---|
| a Mn,theo = ([M]0/[MBP]0) × conv. × MWM. b Experiments B4 and B5 were conducted using outlet polymer solution from experiment T1 as explained in the synthesis procedures. | ||||||
| B1 | 100:1:0.01:0 | 180 | 73 | 6320 | 8140 | 1.28 |
| B2 | 100:1:0.01:0.01 | 180 | 80 | 6880 | 6980 | 3.17 |
| B3 | 100:1:0.01:0.02 | 180 | 79 | 6830 | 7880 | 3.57 |
| B4b | 200:1:0.01:0.01 | 60 | 80 | 13780 | 14050 | 1.16 |
| B5b | 200:1:0.01:0.02 | 60 | 96 | 16520 | 16590 | 1.21 |
As the effects of ascorbic acid in a SET-LRP reaction were not known, it was decided to test the response by starting with a low concentration. Since there was no copper salt added to the system, the ratio was scaled with respect to ligand; experiment B2 and B3 both used a small amount of ascorbic acid, at ratios of 1:1 and 2:1 to Me6TREN. Fig. 4 shows the effect of ascorbic acid on the reaction kinetics. Even with a small amount of ascorbic acid present, there was a boost in polymerization rate and higher conversion was achieved. Unfortunately, ascorbic acid had a detrimental effect on polymerization control. The molecular weight distributions of two samples from experiment B2 are shown in Fig. 5. As the GPC traces show, polymerization was initially uncontrolled and higher molecular weight polymer was formed. As polymerization continued, a low molecular weight distribution was established corresponding to activation and more controlled growth of alkyl halide initiator. Experiment B3 showed similar results, indicating that the presence of ascorbic acid, a strong reducing agent, quickly reduced any copper(II) species initially generated via the SET-LRP catalytic cycle and extinguished control over the polymerization in its early stages.
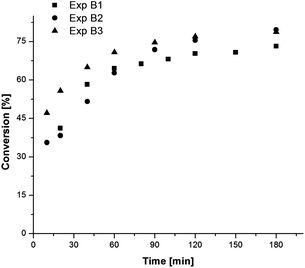 | ||
| Fig. 4 Evolution of conversion as a function of reaction time for batch experiments of methyl acrylate with copper wire and ascorbic acid. Experimental conditions are summarized in Table 2. | ||
 | ||
| Fig. 5 Gel permeation chromatography traces for poly(methyl acrylate) from experiment B2. The traces are normalized by area, and experimental conditions of the experiment are shown in Table 2. | ||
While ascorbic acid added before the SET-LRP catalytic cycle is established has an unfavourable effect over control of the polymerization, the effect is beneficial when added to a polymer solution with existing copper species in the absence of copper wire. The outlet polymer solution from experiment T1 was used to test this effect; the procedure was similar to a chain extension with a small amount of ascorbic acid added to reinitiate polymerization with additional monomer. Experiments B4 and B5 both polymerized rapidly once ascorbic acid was injected, with excellent control over the polymerization. The conversion and molecular weight data for B4 and B5 are shown in Fig. 6 along with that of experiment B1 as a comparison.
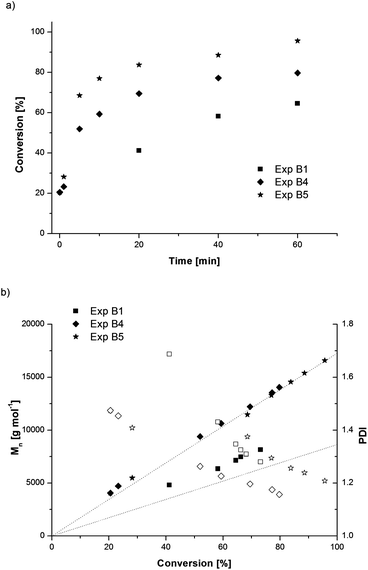 | ||
| Fig. 6 Evolution of conversion vs. time (a), and number-average molecular weight (Mn, filled symbols) and polydispersity index (PDI, open symbols) with conversion (b) for poly(methyl acrylate) prepared by SET-LRP with ascorbic acid at 30 °C. Experimental conditions are summarized in Table 2. | ||
With a one to one ratio of ascorbic acid to ligand, experiment B4 reached 80% conversion in 60 min, while experiment B5, with double the amount of ascorbic acid, reached 95% conversion in the same time. Both experiments showed a linear growth in molecular weight with conversion, falling almost exactly along the theoretical line and almost reaching the target molecular weights. The polydispersity decreased to a final value of 1.16 for experiment B4 and 1.21 for experiment B5. These experiments show that with an existing population of copper(II) species in solution, a small amount of ascorbic acid was sufficient to reduce copper(II) into copper(I) species, which then acts as activators or undergoes the SET-LRP catalytic cycle to generate copper(0) activating species.
Effects of ascorbic acid on SET-LRP in a continuous tubular reactor
By utilizing ascorbic acid and an inert reactor, it is now possible to solve the three challenges originally discussed. The improved design is shown in Fig. 1 in the Experimental section. Rather than constructing the entire reactor out of copper tubing, a short copper coil is used to continuously initiate polymerization and generate soluble copper species. The reaction can then be carried to higher conversion in inert stainless steel tubing, using ascorbic acid fed separately as a reducing agent to drive the activation and deactivation equilibrium. This configuration limits the amount of tubing vulnerable to copper leaching and minimizes the amount of copper in the system. In addition, the residence time and reactor length can now be extended, as the concentration gradients that occur at high conversion in the copper reactor are eliminated in the inert stainless steel tubing as the catalysis is now homogeneous across the radial direction of the reactor.The experimental conditions and average steady state properties of poly(methyl acrylate) polymerized in the new reactor are summarized in Table 3. Experiments T5, T6, and T7 were conducted to test the effect of ascorbic acid concentration on reaction kinetics and polymer properties, with the ascorbic acid level increasing from 1:1, to 2:1, and finally 4:1 with respect to ligand. Note that the feed for the copper reactor was 100:1:0.01 without any added ascorbic acid. Thus, the properties of the polymer exiting the copper tube are similar to those reported for experiment T1 in Table 1. The 40% conversion at the outlet of the copper tube is diluted by the addition of the feed from Section 2 such that the inlet conversion for the stainless steel reactor is 20%, and the target DPn is doubled to 200. All three experiments produced living polymer with high initiator efficiency and predictable molecular weights well in excess of what was possible with only a copper reactor.
| Exp. | [M]0:[MBP]0:[Me6TREN]0:[AscA]0 | Reactor lengtha (m) | τ (min) | convb (%) | Mn,theoc (g mol−1) | Mn,GPC (g mol−1) | PDI | Ieffd (%) |
|---|---|---|---|---|---|---|---|---|
| a Reactor was composed of a short copper tube (3.81 m) and a longer stainless steel section (15.24 m), details as described in the Experimental section. b Conversion and molecular weight data taken as average steady state values (±standard deviation) after 3 residence times. c Mn,theo = ([M]0/[MBP]0) × conv. × MWM. d Ieff = Mn,theo/Mn,GPC × 100% as calculated from steady state averages. e Experiment T8 conducted without stainless steel tubing after the mixing junction. f Experiment T9 utilized a short copper coil (3.81 m) and two sections of stainless steel tubing (30.48 m). | ||||||||
| T5 | 200:1:0.01:0.01 | 19.05 | 35 | 65 ± 1.9 | 11240 | 12230 ± 350 | 1.45 ± 0.08 | 92 |
| T6 | 200:1:0.01:0.02 | 19.05 | 35 | 65 ± 1.3 | 11180 | 11800 ± 260 | 1.47 ± 0.05 | 95 |
| T7 | 200:1:0.01:0.04 | 19.05 | 35 | 67 ± 1.6 | 11550 | 11800 ± 440 | 1.42 ± 0.03 | 98 |
| T8e | 200:1:0.01:0.02 | 3.81 | 8 | 27 ± 1.3 | 4650 | 4670 ± 220 | 1.43 ± 0.03 | 99 |
| T9f | 200:1:0.01:0.02 | 34.29 | 62 | 78 ± 1.1 | 13500 | 13840 ± 380 | 1.27 ± 0.03 | 98 |
Conversion, Mn, and PDI as a function of dimensionless residence time (t/τ) for experiments T5–T7 are shown in Fig. 7. The steady state values for conversion and Mn are 65%/12230 g mol−1, 65%/11800 g mol−1, and 67%/11800 g mol−1 for ratios of 1:1, 2:1 and 4:1 ascorbic acid to ligand, respectively. The steady state conversion and molecular weight for all three experiments were approximately equal, indicating that for these conditions additional ascorbic acid had a negligible effect on reaction kinetics. Conversion and number average molecular weight were relatively stable over several residence times.
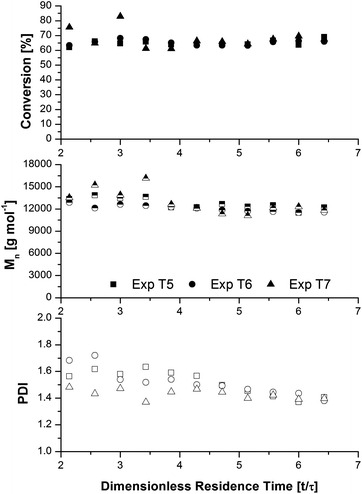 | ||
| Fig. 7 Evolution of conversion (closed symbols), number-average molecular weight (half-filled symbols), and polydispersity index (open symbols) as a function of dimensionless residence time for methyl acrylate SET-LRP polymerizations at ambient temperature in a copper tubular reactor with ascorbic acid at a residence time of 35 min. Experimental conditions are summarized in Table 3. | ||
A peculiar observation was the slow decrease in PDI with increasing dimensionless residence time for all three experiments. Since Mn remained relatively stable, the downwards drift in PDI indicated a decrease in the weight average molecular weight. One possible explanation for this behaviour is poor mixing in the junction where the two feeds meet. A schematic of the mixing junction, constructed using a Tee union and a 10 cm piece of stainless steel tubing with a total volume of approximately 4 mL, is shown in Fig. 8. The simple flow through design is intended to provide additional mixing time for the two feeds to equilibrate before the polymer solution entered the stainless steel reactor. However, without active agitation the mixing behaviour is most likely non-ideal, leading to a broadening of the residence time distribution. Any polymer build-up in the dead space would lead to a gradual elution of higher molecular weight polymer until the reactants in the mixing junction become almost homogeneous. The effect of non-ideal flow behaviour has been modelled for reversible addition-fragmentation chain transfer (RAFT) polymerization in a continuous flow reactor by Bitsch et al., and shown to have a significant impact on the resulting molecular weight distribution.47
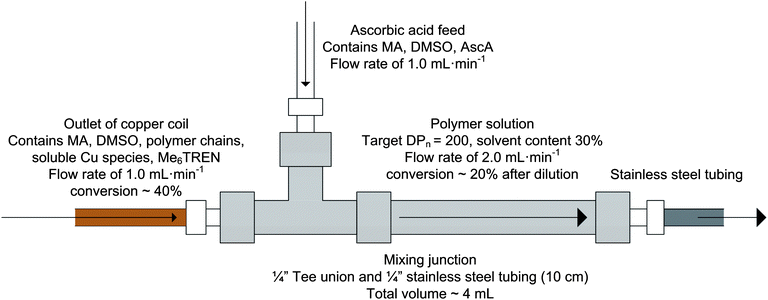 | ||
| Fig. 8 Schematic of mixing junction between the outlet of the copper reactor, the inlet of the ascorbic acid feed and the stainless steel reactor. | ||
To test this theory, an experiment (T8) was conducted to sample the material exiting the mixing junction. Conversion, Mn, and PDI as a function of reaction time for experiment T6 (exit of stainless steel reactor) and T8 (exit of mixing junction) are shown in Fig. 9. The first sample at time 0 of experiment T8 was taken before ascorbic acid was fed into the mixing chamber to confirm that the conversion out of the copper coils was approximately 40% as observed in experiment T1. Once the ascorbic acid feed was started, the reaction mixture was diluted and steady state conversion stabilized at approximately 27%, with an Mn of 4670 g mol−1. Although the PDI drift was not as significant as in experiment T6, the PDI of T8 decreased from 1.45–1.50 to 1.40 after approximately 150 min, providing support for poor mixing in the junction as a cause of PDI drift.
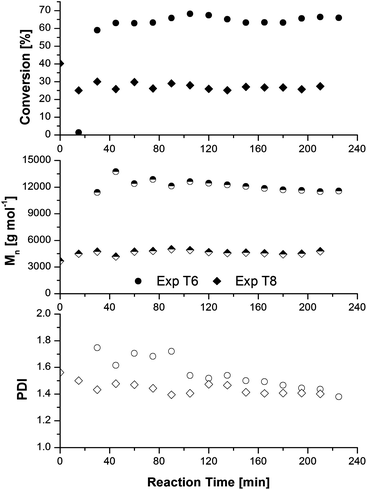 | ||
| Fig. 9 Evolution of conversion (closed symbols), number-average molecular weight (half-filled symbols), and polydispersity index (open symbols) as a function of reaction time for poly(methyl acrylate) samples at the outlet of the mixing junction and the outlet of the stainless steel reactor. Experimental conditions are summarized in Table 3. | ||
Since increasing the concentration of ascorbic acid had little effect on steady state conversion values, the residence time was increased by adding another section of stainless steel tubing to give a total residence time of 62 min (experiment T9). Fig. 10 shows the evolution of conversion, Mn, and PDI for experiment T6 and T9 as a function of dimensionless residence time (t/τ). As expected, increasing the residence time led to an increase in conversion to 78% along with an increased Mn of 13840 g mol−1 and a decrease of PDI to 1.27. The narrower molecular weight distribution can be attributed to the higher outlet conversion and a corresponding larger number of activation/deactivation cycles. The results from experiment T9 demonstrate that ascorbic acid can be used as reducing agent to drive a controlled SET-LRP polymerization to high conversion in the absence of copper surface. The use of stainless steel tubing to construct a continuous tubular reactor was validated, and the proposed reactor design could be used as a continuous process to produce well controlled and living polymer chains. Although methyl acrylate was used as monomer in both feeds in the study, a second monomer could have been used in the second feed to produce a block gradient copolymer instead. One can readily envision the use of a water soluble monomer such as N-isopropylacrylamide in the first feed to produce an amphiphilic block/gradient copolymer in one continuous process.
 | ||
| Fig. 10 Evolution of conversion (closed symbols), number-average molecular weight (half-filled symbols), and polydispersity index (open symbols) as a function of dimensionless residence time for methyl acrylate SET-LRP polymerizations at ambient temperature in a copper tubular reactor with ascorbic acid at residence times of 35 and 62 min. Experimental conditions are summarized in Table 3. | ||
Chain extension of outlet polymer
Although ascorbic acid was shown to work well as a reducing agent, it was necessary to keep the ascorbic acid flow separate from ligand until the start of polymerization, as the basic nitrogen based ligand was prone to protonation by ascorbic acid. Additionally, during the reduction process, ascorbic acid was presumably oxidized to form dehydroascorbic acid as well as two quantities of hydrogen halide.48,49 Since the initiator contained bromine, the reduction process would have formed hydrogen bromide or hydrobromic acid. As the products of the reduction process may also have protonated the ligand, the series of reactions may have been detrimental to retention of chain end functionality. To demonstrate the livingness of the polymer produced from the tubular reactor, chain extensions of the outlet polymer solutions from experiments T5–T9 were conducted with additional monomer. Although other monomers could have been used to produce a block copolymer, for simplicity methyl acrylate was again used to simulate the additional block. The results of the chain extension are summarized in Table 4.| Exp. | [M]0:[MBP]0:[Me6TREN]0:[AscA]0a | convinitb (%) | Mn, init (g mol−1) | PDIinit | convfinal (%) | Mn, final (g mol−1) | PDIfinal |
|---|---|---|---|---|---|---|---|
| a Concentrations of Me6TREN and AscA shown are for reactants added during the chain extension procedure and exclude the concentrations initially present in the polymer solution. b Initial conversion after dilution from monomer and solvent addition. | |||||||
| X1 | Ext. of T5, 400:1:0:0.01 | 30 | 11200 | 1.46 | 31 | 11200 | 1.46 |
| X2 | Ext. of T5, 400:1:0.02:0.01 | 30 | 11200 | 1.46 | 88 | 28990 | 1.22 |
| X3 | Ext. of T6, 400:1:0.04:0.01 | 43 | 13800 | 1.38 | 88 | 27810 | 1.19 |
| X4 | Ext. of T7, 400:1:0.08:0.01 | 45 | 15320 | 1.35 | 90 | 29390 | 1.19 |
| X5 | Ext. of T9, 400:1:0.04:0.01 | 44 | 14200 | 1.30 | 91 | 25750 | 1.20 |
The chain extension experiments were performed using outlet polymer solution that had been exposed to air for greater than 48 h. The first chain extension X1 was prepared with no additional ligand. The injection of ascorbic acid to reinitiate polymerization was unsuccessful, as the remaining ligand was likely protonated by residual acid generated in the reduction process. Chain extension of the same polymer solution was performed with the addition of a small amount of ligand and fresh ascorbic acid (X2). Supplemental ligand was added in a 2 fold excess to the amount of ascorbic acid that would have been present in the solution from the original polymerization. The additional ligand was able to complex to existing copper species and displaced any protonated ligand to form active complexes. Upon injection of fresh ascorbic acid, the polymerization was successfully reinitiated and the conversion increased to 88% in 90 min. The Mn increased from 11200 to 28990 g mol−1, and the PDI decreased from 1.46 to 1.22. Chain extensions of the other outlet polymer solutions (X3–X5) were likewise successful, as all the experiments exhibited a clear growth in molecular weight with increasing conversion in a short period of time. The GPC traces of chain extension X2 are plotted along with a molecular weight distribution from experiment T8 (outlet of copper coil) in Fig. 11. The traces are normalized with respect to area, and show a noticeable narrowing of the molecular weight distribution upon extension, as well as a clean shift to higher molecular weight values.
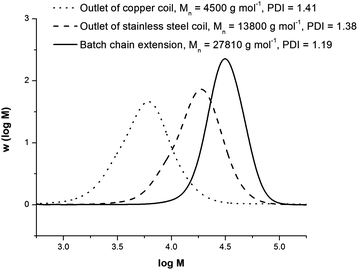 | ||
| Fig. 11 Gel permeation chromatography traces for pMA chain extensions of experiment T6. The traces are normalized by area, and experimental conditions of the extensions are shown in Table 4. | ||
These chain extensions show that the polymer chains produced are highly living, and that storage with ascorbic acid (and by-products from the reduction process) for an extended period of time had a negligible impact on the chain end functionality. Although a small amount of ligand had to be added to re-initiate polymerization, no additional copper was added to the solution. The copper concentrations in the polymer solutions coming out of the stainless steel reactor were measured by flame atomic absorption spectroscopy and found to be approximately 15–20 ppm. The values were in agreement with those found in a previous SET-LRP study utilizing copper wire as a catalyst source in a continuous stirred tank reactor.50 With the additional dilution by subsequent monomer addition, the chain extensions were mediated by less than 10 ppm of copper in solution. At such a low copper concentration, the final polymer may be usable in a variety of applications without further purification. With two reactor feeds and the subsequent chain extension, the methodology could be used to produce a triblock gradient copolymer. Conceptually the chain extension could also be added to the continuous process with additional feed points and stainless steel tubing.
Conclusions
An innovative design for a flow reactor was proposed for the continuous production of uniform polymer with high livingness using SET-LRP, improving upon the initial concept.28 Instead of using copper tubing to construct the entire reactor, a short copper coil was used to initiate polymerization and generate soluble copper species. The bulk of the reaction then took place in inert stainless steel tubing, using ascorbic acid as a reducing agent to drive the catalytic cycle and mediate the polymerization. The study is the first investigation into the use of ascorbic acid in SET-LRP reactions. The addition of ascorbic acid into a batch reaction with copper wire gave uncontrolled polymerization, likely due to very fast reduction of copper(II) generated at the onset of polymerization such that insufficient deactivators were in solution. However, when a small amount of ascorbic acid was added to a polymer solution with soluble copper species, such as the material exiting the copper tubing, the ascorbic acid was able to regenerate active copper(I) and copper(0) species to re-initiate polymerization in the absence of copper(0) surface.The improved tubular system was able to continuously polymerize methyl acrylate at ambient temperature with 30 wt% DMSO as solvent, reaching a steady state conversion of 65–67% with a residence time of 35 min. The residence time was extended to 62 min by adding another length of stainless steel tubing. Using this reactor configuration, it was possible to reach a steady state conversion of 78% and produce well defined living polymer with a narrow molecular weight distribution.
Chain extension experiments were conducted on the outlet polymer solution. It was found that additional ligand was required to re-initiate polymerization, likely due to protonation of ligand already existing in the polymer solution by residual ascorbic acid and the derivatives generated in the reduction process. With a small excess of ligand, the chain extensions were well-controlled and showed a clean shift and narrowing in molecular weight distribution with increasing conversion. Although methyl acrylate was used in all stages in this study to simplify analysis, other monomers could have been substituted at any of the feed points or in the chain extension to generate triblock gradient copolymers. The chain extension could also be easily adapted to the continuous reactor by the addition of two feed points, and additional stainless steel tubing. The results illustrate the significant potential of using a continuous tubular reactor as an efficient means to scale-up production of well controlled polyacrylics and other multiblock copolymers.
Notes and references
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- J. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5098–5112 CrossRef CAS.
- Y. Shen, H. Tang and S. Ding, Prog. Polym. Sci., 2004, 29, 1053–1078 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS.
- L. Mueller and K. Matyjaszewski, Macromol. React. Eng., 2010, 4, 180–185 CrossRef CAS.
- M. Destarac, Macromol. React. Eng., 2010, 4, 165–179 CrossRef CAS.
- W. Jakubowski, K. Min and K. Matyjaszewski, Macromolecules, 2006, 39, 39–45 CrossRef CAS.
- K. Matyjaszewski, W. Jakubowski, K. Min, W. Tang, J. Huang, W. a Braunecker and N. V. Tsarevsky, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15309–15314 CrossRef CAS.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS.
- G. Lligadas, B. M. Rosen, M. J. Monteiro and V. Percec, Macromolecules, 2008, 41, 8360–8364 CrossRef CAS.
- G. Lligadas, B. M. Rosen, C. A. Bell, M. J. Monteiro and V. Percec, Macromolecules, 2008, 41, 8365–8371 CrossRef CAS.
- X. Jiang, S. Fleischmann, N. H. Nguyen, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5591–5605 CrossRef CAS.
- M. J. Monteiro, T. Guliashvili and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1835–1847 CrossRef CAS.
- Y. Zhang, Y. Wang and K. Matyjaszewski, Macromolecules, 2011, 44, 683–685 CrossRef CAS.
- A. G. West, B. Hornby, J. Tom, V. Ladmiral, S. Harrisson and S. Perrier, Macromolecules, 2011, 44, 8034–8041 CrossRef CAS.
- Y. Zhang, Y. Wang, C.-H. Peng, M. Zhong, W. Zhu, D. Konkolewicz and K. Matyjaszewski, Macromolecules, 2012, 45, 78–86 CrossRef CAS.
- K. Min, W. Jakubowski and K. Matyjaszewski, Macromol. Rapid Commun., 2006, 27, 594–598 CrossRef CAS.
- K. Matyjaszewski, H. Dong, W. Jakubowski, J. Pietrasik and A. Kusumo, Langmuir, 2007, 23, 4528–4531 CrossRef CAS.
- H. Tang, Y. Shen, B.-G. Li and M. Radosz, Macromol. Rapid Commun., 2008, 29, 1834–1838 CrossRef CAS.
- N. Chan, S. Boutti, M. F. Cunningham and R. A. Hutchinson, Macromol. React. Eng., 2009, 3, 222–231 CrossRef CAS.
- S. Fleischmann, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1190–1196 CrossRef CAS.
- S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2243–2250 CrossRef CAS.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5109–5119 CrossRef CAS.
- N. Chan, M. F. Cunningham and R. A. Hutchinson, Macromol. Rapid Commun., 2011, 32, 604–609 CrossRef CAS.
- K. Matyjaszewski, N. V. Tsarevsky, W. A. Braunecker, H. Dong, J. Huang, W. Jakubowski, Y. Kwak, R. Nicolay, W. Tang and J. A. Yoon, Macromolecules, 2007, 40, 7795–7806 CrossRef CAS.
- A. A Isse, A. Gennaro, C. Y. Lin, J. L. Hodgson, M. L. Coote and T. Guliashvili, J. Am. Chem. Soc., 2011, 133, 6254–6264 CrossRef.
- N. H. Nguyen, B. M. Rosen, G. Lligadas and V. Percec, Macromolecules, 2009, 42, 2379–2386 CrossRef CAS.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS.
- M. E. Levere, I. Willoughby, S. O'Donohue, A. de Cuendias, A. J. Grice, C. Fidge, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1086–1094 RSC.
- Y. Shen, S. Zhu and R. Pelton, Macromol. Rapid Commun., 2000, 21, 956–959 CrossRef CAS.
- Y. Shen and S. Zhu, AIChE J., 2002, 48, 2609–2619 CrossRef CAS.
- M. Müller, M. F. Cunningham and R. A. Hutchinson, Macromol. React. Eng., 2008, 2, 31–36 CrossRef.
- T. Noda, A. J. Grice, M. E. Levere and D. M. Haddleton, Eur. Polym. J., 2007, 43, 2321–2330 CrossRef CAS.
- H. Chen, M. Zhang, M. Yu and H. Jiang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4721–4724 CrossRef CAS.
- G. J. P. Britovsek, J. England and A. J. P. White, Inorg. Chem., 2005, 44, 8125–8134 CrossRef CAS.
- T. Gruendling, T. Junkers, M. Guilhaus and C. Barner-Kowollik, Macromol. Chem. Phys., 2010, 211, 520–528 CrossRef CAS.
- N. H. Nguyen, X. Jiang, S. Fleischmann, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5629–5638 CrossRef CAS.
- F. Stoffelbach, N. Griffete, C. Bui and B. Charleux, Chem. Commun., 2008, 4807–4809 RSC.
- J. K. Oh, K. Min and K. Matyjaszewski, Macromolecules, 2006, 39, 3161–3167 CrossRef CAS.
- L. A. McCullough, B. Dufour and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5386–5396 CrossRef CAS.
- K. Min, H. Gao and K. Matyjaszewski, Macromolecules, 2007, 40, 1789–1791 CrossRef CAS.
- S. M. Paterson, D. H. Brown, T. V. Chirila, I. Keen, A. K. Whittaker and M. V. Baker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4084–4092 CrossRef CAS.
- B. Bitsch, C. Barner-Kowollik and S. Zhu, Macromol. React. Eng., 2011, 5, 55–68 CrossRef CAS.
- M. J. W. Taylor, W. T. Eckenhoff and T. Pintauer, Dalton Trans., 2010, 39, 11475–11482 RSC.
- R. Casolari, F. Felluga, V. Frenna, F. Ghelfi, U. M. Pagnoni, A. F. Parsons and D. Spinelli, Tetrahedron, 2011, 67, 408–416 CrossRef CAS.
- N. Chan, M. F. Cunningham and R. A. Hutchinson, Polym. Chem., 2012, 3, 486–497 RSC.
| This journal is © The Royal Society of Chemistry 2012 |