 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A general solid phase method for the synthesis of depsipeptides†
Mary M.
Nguyen
,
Nicole
Ong
and
Laura
Suggs
*
The University of Texas at Austin, Department of Biomedical Engineering, 107 W Dean Keeton Street, Austin, TX 78712, USA. E-mail: m.nguyen@utexas.edu; nicoleong@utexas.edu; Laura.Suggs@engr.utexas.edu; Fax: +1 512 471 0616; Tel: +1 512 232 8593
First published on 2nd January 2013
Abstract
Herein we describe the synthesis of depsipeptide sequences in which the backbone is composed of alternating esters and amides. Our methodology is based on the synthesis and protection of a depsidipeptide block, which is used as the growing unit for manual SPPS. We have explored Fmoc/OBzl and Fmoc/tBu SPPS strategies, and found the latter to be most compatible with our methodology.
Introduction
Depsipeptides are an interesting class of molecules that incorporate esters within a peptide backbone. Depsipeptides are readily found in nature, for example the marine depsipeptide families of didemnins are isolated from Trididemnum solidum and play an important role in the defense mechanisms of marine natural products.1 The synthesis of depsipeptides has previously been explored using solid phase peptide synthesis (SPPS). Kuilse et al. coupled α-hydroxy acid protected lactic acid and mandelic acid to Boc-protected α-amino acids on a Wang resin using DIC and DMAP.2 Spengler et al. devised a machine-assisted protocol for a family of depsipeptides based on a sequence of 26 residues with up to 6 ester substitutions.3 The highest yields (∼30%) of the final product were seen with 1 or 2 ester substitutions, while substitutions of 6 esters gave relatively low yields (7%). Both bodies of work have shown that single and multiple esters are successfully incorporated into a peptide backbone without modification of traditional SPPS methods. Ester substitutions of peptide sequences are a common strategy to analyze protein folding, function, and self-assembly.4,5 Protein folding and self-assembly is governed by a variety of supramolecular interactions, such as hydrogen-bonding, pi–pi, electrostatic, or hydrophobic interactions between peptides, peptide side chains, and/or protecting groups. Despite the reduction of hydrogen bonding interactions, depsipeptides are shown to have a stronger propensity to form α-helices rather than β-sheets, as seen with Leu-Leu-Lac-OEt repeats.6 Hydrogel formation from depsipeptides of amyloid derivatives was dependent on the number and location of the ester substitutions within the 10 residue sequence.4 Depsipeptides with three ester substitutions yielded gel fibers that resembled large helical ribbons while sequences with 1 ester substitution did not form a gel. A computational, molecular-mechanics model of a 12 residue, alternating sequence of lactic acid and lysine show the potential for regular folding patterns for depsipeptides with alternating ester and amide bonds.7 Limited work has been focused on the synthesis of depsipeptides with regular repeats of esters. Here, we propose a synthetic methodology in which the depsipeptide sequence has regular, alternating esters between charged peptide residues.Results and discussion
A general strategy towards the synthesis of depsipeptides includes the synthesis of unique building blocks, and incorporating the building blocks into a peptide chain via traditional methods.8 Using the above strategy, we have synthesized depsipeptides of varying lengths and sequences (Fig. 1) using solid phase peptide synthesis (SPPS). We designed our depsidipeptides with a Fmoc-protected N-terminus, lactic acid (Lac) as the ester moiety to maintain hydrophobicity, and either lysine (Lys) or aspartic acid (Asp) as the charged entities. Our system involves the synthesis of Fmoc-depsidipeptides “building blocks” (Scheme 1). Synthesizing the building blocks for SPPS requires protection at the N- and C-terminus, as well as the peptide side chain. While a number of protected peptides are commercially available, protecting a depsipeptide building block is not as straightforward. Removal of base-labile groups has been shown to affect the stereochemistry of the ester bond2 while deprotection of acid-labile groups may hydrolyze the ester bond. The synthesis of Fmoc-dipeptides has been reported with O-pentafluorophenol (Pfp)-activated esters,9 which motivated us to take a similar strategy. We synthesized the O-Pfp esters of Fmoc-Lys(Z)-OH and Fmoc-Asp(OBzl)-OH then coupled them to lactic acid with DIPEA in DCM. Our initial strategy was to use the Fmoc/OBzl strategy for SPPS with these depsidipeptides, limiting the acidic exposure to the last step of the reaction, i.e. cleaving from the resin. The coupling reactions were monitored via TLC. The samples were filtered and concentrated, and extracted with ethyl acetate.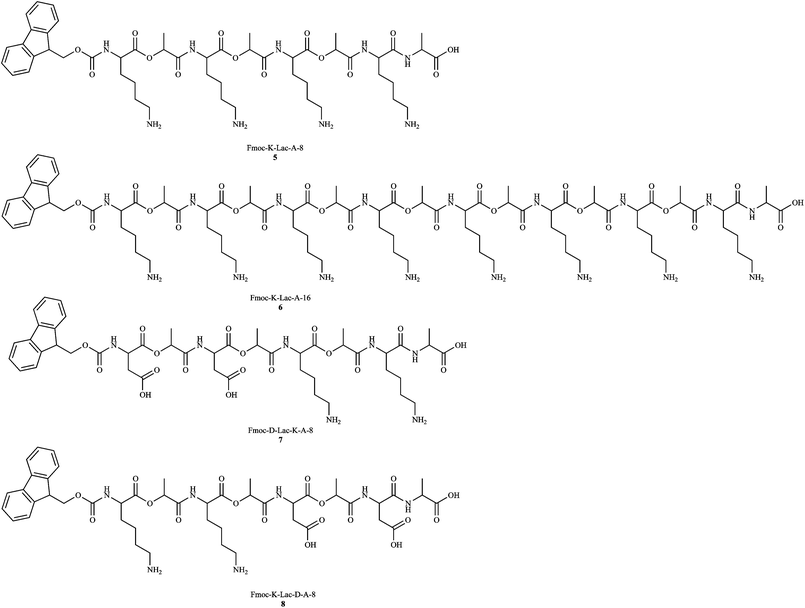 | ||
| Fig. 1 Depsipeptide library: we have synthesized depsipeptides 5–8 with alternating esters and charged peptides. All of the sequences are protected at the N-terminal with an Fmoc group and are capped at the C-terminal with alanine (A). The sequences are named according to the residue (K = lysine, D = aspartic acid, Lac = lactic acid) and total number of residues. | ||
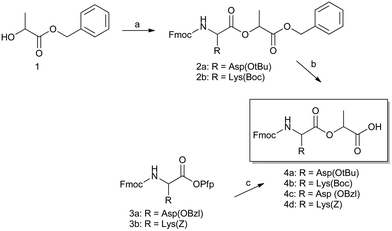 | ||
| Scheme 1 Fmoc-depsidipeptide synthesis, whereby (a) Fmoc-peptide in DCM, DIC/DMAP, 0 °C for 1 h, 16 hours at room temperature. Silica column in hexanes and ethyl acetate (67–82%); (b) Pd/C in H2 at 5 or 15 psi for up to 16 h in dry methanol, silica column in DCM and methanol (33–67%); (c) lactic acid in DCM with DIPEA 0 °C for 1 h, 16 hours at room temperature. Silica column in hexanes and ethyl acetate. The synthesis of structures 110 and 39 have been described elsewhere. | ||
Upon purification via silica column chromatography in either hexanes/ethyl acetate or DCM/methanol/acetic acid, we found that Fmoc-peptide-OH remained in the sample mixtures. Purification by recrystallization was not successful.
We proceeded to protect the α-hydroxy acid with a benzyl group.10 Lac was dissolved in ethyl acetate with benzyl chloride and TEA. The mixture was refluxed for 5 hours, and the desired product was subsequently purified via distillation. 1H-NMR (not shown) matched that reported in the literature.10 Protections with the benzyl group required us to change the original protection of the peptides used in the OPfp-activation methods. With this strategy, we worked with OtBu- and Boc-protected Fmoc-peptides, specifically coupling (1) with Fmoc-Lys(Boc)-OH and Fmoc-Asp(OtBu)-OH in DCM with DIC and DMAP. The reactions were monitored by TLC. The samples were filtered, reduced, and purified on silica in hexanes and ethyl acetate. The benzyl-protecting group was removed in dry methanol with 10% palladium on activated carbon under hydrogen and monitored with TLC. The deprotected sample was purified on a silica column in DCM and methanol and confirmed to be analytically pure with NMR and MS/LCMS. The absence of broad peaks in the 1H-NMR spectrum suggests the Fmoc-depsidipeptides are not racemized under the reaction conditions we used.
Our depsipeptides are designed to have regularly alternating esters and amides within their backbone. We have synthesized both charged and self-complementary sequences using SPPS with Lys, Asp, Ala, and Lac residues. SPPS proceeded with standard Fmoc strategies on a trityl chloride resin (Scheme 2) and was monitored with ninhydrin. Preliminary tests were conducted with crude samples of 4d as the building block, to ensure the depsipeptide was stable under general SPPS conditions. The resin was coupled initially with Lac and DIPEA in DCM for 2 hours and washed with DCM. Then Fmoc-Lys(Z)-OH was coupled with DIC and DMAP and washed with DMF and DCM. The depsidipeptide was coupled with DIC and Oxymapure 3 times and cleaved in 1% TFA in DCM, with washings in between each coupling step. ESI/MS reveals that we were successful in synthesizing Fmoc-octodepsipeptide, however a peak originating from the Fmoc-septadepsipeptide was observed. Further analysis with fragmented MS showed that Lac at the C-terminal was hydrolyzed. We also observed that the internal esters were not disrupted during any of the SPPS methods nor were they ionized during the analysis. To prevent hydrolysis of Lac at the C-terminal upon cleaving, Fmoc-Ala-OH was used for the first coupling step. Of the four Fmoc-depsidipeptides synthesized via our methodology, (4a) and (4b) were successfully purified, and we proceeded to use them with our optimized SPPS protocol using Fmoc/OtBu strategies. This new strategy required us to optimize the cleaving cocktail, as the mixture needed to simultaneously cleave the sequence off the resin and remove the Boc or OtBu protecting groups without hydrolyzing the ester bonds.
 | ||
| Scheme 2 Synthesis of oligodepsipeptides via standard Fmoc-SPPS methods with a trityl chloride resin, whereby (a) DIPEA (1 equiv.) for 1.5 hours with mixing, followed by DMF and DCM washes; (b) 20% piperidine in DMF for 5 minutes (×3) followed by DMF and DCM washes; (c) DIC (4 equiv.) and DMAP (0.01 equiv.) for 2 hours with mixing, followed by DMF and DCM washes; (d) DIC (4 equiv.) and OxymaPure (0.1 equiv.) for 2 hours with mixing followed by DCM washes; (e) mixing with cleaving cocktail (A: TFA/TIPS – 95/5; B: TFA/Water/TIPS – 95/2/3; C: TFA/DCM/TIPS – 95/2/3; D: TFA/DCM/TIPS – 50/48/2) for 3 hours followed by precipitation in cold ether or extraction in chloroform. | ||
We tested four standard mixtures on Fmoc-K-Lac-8: A: TFA/TIPS – 95/5; B: TFA/Water/TIPS – 95/2/3; C: TFA/DCM/TIPS – 95/2/3; D: TFA/DCM/TIPS – 50/48/2. Upon mixing for 3 hours, the samples were precipitated in cold ether, cooled overnight, and centrifuged. MALDI of the crude samples were very similar among all of the cocktails and also suggest minimal hydrolysis of the esters in all cocktails. We also synthesized a 16-mer with (4b), which was cleaved with mixture B. Again, no internal esters were hydrolyzed, suggesting that longer sequences can successfully be synthesized with our methodology. We attempted to synthesize Fmoc-D-Lac-8 and Fmoc-D-Lac-6. Both sequences did not precipitate well in ether upon cleavage and were extracted in chloroform prior to purification with HPLC. While ESI/MS (not shown) confirmed that both were synthesized, neither sequence was successfully purified.
SPPS on the trityl chloride resin gave very low yields (2–3%). We proceeded to use a Fmoc-Ala-Wang resin and used the synthesis methods as outlined in Scheme 2 with the exception of eliminating the first coupling step with Lac (a). Under the same Fmoc/OtBu strategies as described above, yields increased to 30–40%. We proceeded to use Fmoc-Ala-Wang resin for the synthesis of ionic, self-complementary depsipeptides Fmoc-K-Lac-D-A-8 and Fmoc-D-Lac-K-A-8, showing additional evidence that our method can be used for a variety of depsipeptide sequences.
Conclusions
We have successfully synthesized depsipeptides with alternating esters and amides of varying lengths and sequences. Our SPPS method involves the synthesis of unique Fmoc-depsidipeptides as the building block. Our strategy allows for scale-up of the synthesis and can be used as a template to incorporate α-hydroxy acids and peptide residues not explored in this work. The sequences purified in this article will be further explored for their tendency to self-assemble and the results will be the subject of a future publication.Acknowledgements
This work was supported by the Welch Foundation and the Nation Institutes of Health (R21HL102806). We would like to thank Cameron Peebles and Dr John Hardy for productive discussions and for their time in revising the manuscript. Also many thanks to the NMR Characterization Facility staff, Dr Ben Shoulders, Steve Sorey, and Angela Spangelberg; the MS Facility and Karin Keller; and the Protein and Metabolite Analysis Facility staff, Michelle Gadush and Marvin Mercado. We would also like to acknowledge Dr Brent Iverson and William Bell for their critical input.Notes and references
- J. Lee, J. N. Currano, P. J. Carroll and M. M. Joullie, Nat. Prod. Rep., 2012, 29(3), 404–424 RSC.
- O. Kuisle, E. Quinoa and R. Riguera, J. Org. Chem., 1999, 64(22), 8063–8075 CrossRef CAS.
- J. Spengler, B. Koksch and F. Albericio, Biopolymers, 2007, 88(6), 823–828 Search PubMed.
- R. C. Elgersma, T. Meijneke, G. Posthuma, D. T. S. Rijkers and R. M. J. Liskamp, Chem.–Eur. J., 2006, 12(14), 3714–3725 CrossRef CAS.
- X. Y. Yang, M. Wang and M. C. Fitzgerald, Bioorg. Chem., 2004, 32(5), 438–449 Search PubMed.
- H. Oku, K. Yamada and R. Katakai, Biopolymers, 2008, 89(4), 270–283 Search PubMed.
- J. J. Zhang, M. King, L. Suggs and P. Y. Ren, Biomacromolecules, 2007, 8(10), 3015–3024 Search PubMed.
- M. Stawikowski and P. Cudic, Depsipeptide synthesis, in Methods in Molecular Biology, ed. G. B. Fields, 2007, vol. 386, pp. 321–339 Search PubMed.
- L. Kisfaludy and I. Schon, Synthesis-Stuttgart, 1983,(4), 325–327 Search PubMed.
- W. Fan, X. Li, S. Qian, S. Wang and Y. Wu, Lett. Drug Des. Discovery, 2009, 6(7), 542–547 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods for Fmoc-depsidipeptides 3a, 3b, 4c, and 4d and depsipeptides 5–9. 1H-, 13C-NMR, and MS/LCMS spectra for Fmoc-depsidipeptides 3a, 3b, 4c, and 4d. HPLC spectra for depsipeptides 5–8. See DOI: 10.1039/c2ob26893k |
| This journal is © The Royal Society of Chemistry 2013 |