 Open Access Article
Open Access ArticlePyridostatin analogues promote telomere dysfunction and long-term growth inhibition in human cancer cells†
Sebastian
Müller
a,
Deborah A.
Sanders
a,
Marco
Di Antonio
a,
Stephanos
Matsis
a,
Jean-François
Riou
b,
Raphaël
Rodriguez
a and
Shankar
Balasubramanian
acd
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: rr324@cam.ac.uk; sb10031@cam.ac.uk; Tel: +44 (0)1223 336347
bRegulation et Dynamique des Genomes, Museum National d'Histoire Naturelle, INSERM U565, CNRS UMR 7196, Paris, France
cCancer Research UK, Cambridge Research Institute, Li Ka Shing Center, Cambridge, CB2 0RE, UK
dSchool of Clinical Medicine, University of Cambridge, Cambridge, CB2 0SP, UK
First published on 21st June 2012
Abstract
The synthesis, biophysical and biological evaluation of a series of G-quadruplex interacting small molecules based on a N,N′-bis(quinolinyl)pyridine-2,6-dicarboxamide scaffold is described. The synthetic analogues were evaluated for their ability to stabilize telomeric G-quadruplex DNA, some of which showed very high stabilization potential associated with high selectivity over double-stranded DNA. The compounds exhibited growth arrest of cancer cells with detectable selectivity over normal cells. Long-time growth arrest was accompanied by senescence, where telomeric dysfunction is a predominant mechanism together with the accumulation of restricted DNA damage sites in the genome. Our data emphasize the potential of a senescence-mediated anticancer therapy through the use of G-quadruplex targeting small molecules based on the molecular framework of pyridostatin.
Introduction
Nucleic acids can adopt various non-Watson–Crick secondary structures including G-quadruplexes, a supramolecular architecture that can arise from certain G-rich sequences. Such structures comprise Hoogsteen hydrogen bonded guanines leading to the formation of G-quartets that stack via π–π interactions. The relative orientation of the strands and loop configurations can vary, giving rise to polymorphic conformations that can depend on the primary nucleic acid sequence, temperature, solvent and salt composition.1–4There has been experimental data in support of G-quadruplex formation in the genomic DNA of several organisms.5–8 Lipps, Rhodes and co-workers provided the first strong evidence for their existence in the ciliate Stylonychia. In their report, the authors demonstrated that folding into these motifs at the telomeric region of chromosomes is regulated by the Telomere End Binding Proteins (TEBP) α and β in a cell cycle-dependent manner, and might act as telomere capping structures.9 Human telomeres comprise a highly repetitive G-rich DNA repeat sequence (TTAGGG)n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 and the human telomeric G-quadruplex has been extensively studied in vitro.11,12 It has been postulated that these motifs affect telomere elongation,13 capping14 and replication in vivo.15 The production of telomeric repeat-containing RNA (TERRA) may also suggest a role during transcription.16 In a seminal paper, Zahler et al. showed that certain G-rich telomeric sequences can form G-quadruplexes in vitro, rendering them resistant to extension by the reverse transcriptase telomerase.17 Successive rounds of replication leads to progressive telomere shortening in normal cells, an event that is alleviated by telomerase conferring infinite proliferative capacity in ∼85% of cancer cells.18 These findings prompted us to design selective G-quadruplex interacting small molecules with the aim to develop a novel telomere based anticancer therapy.19 However, recent reports have suggested that such molecules can induce telomere dysfunction in a telomerase independent manner.14,20,21
10 and the human telomeric G-quadruplex has been extensively studied in vitro.11,12 It has been postulated that these motifs affect telomere elongation,13 capping14 and replication in vivo.15 The production of telomeric repeat-containing RNA (TERRA) may also suggest a role during transcription.16 In a seminal paper, Zahler et al. showed that certain G-rich telomeric sequences can form G-quadruplexes in vitro, rendering them resistant to extension by the reverse transcriptase telomerase.17 Successive rounds of replication leads to progressive telomere shortening in normal cells, an event that is alleviated by telomerase conferring infinite proliferative capacity in ∼85% of cancer cells.18 These findings prompted us to design selective G-quadruplex interacting small molecules with the aim to develop a novel telomere based anticancer therapy.19 However, recent reports have suggested that such molecules can induce telomere dysfunction in a telomerase independent manner.14,20,21
Telomeres comprise a protein complex named shelterin,22 which covers and protects the ends of telomeres from the DNA-damage response machinery that promote non-homologous end-joining and homologous recombination. Small molecules interacting with G-quadruplexes have been shown to induce telomere dysfunctions by competing for binding with the shelterin components Telomeric Repeat Factor 1 (TRF1), Telomeric Repeat Factor 2 (TRF2) and Protection of Telomeres 1 (POT1), leading to progressive telomere shortening, anaphase bridges and aberrant chromosome segregation.14,15,23 In addition, dysfunctional telomeres have been shown to activate the DNA damage response, resulting in cell cycle checkpoint activation and growth arrest.24 Several putative G-quadruplex sequences, besides the telomeric repeat, have been identified in the human genome25 with an enrichment in promoter regions of several proto-oncogenes26 such as c-kit,27,28c-myc,29,30bcl-231 and K-ras.32 Small molecules have been proposed to interact with promoter quadruplexes and to alter the expression of those genes in cellulo.29,30,33,34 These motifs have also been identified in RNA35–37 where they might have a functional role by modulating translation21,38 and alternative splicing.39 Further evidence for the existence of G-quadruplex motifs in human cells has been provided by recent work from Gibbons and co-workers, demonstrating that the alpha thalassemia/mental retardation syndrome X-linked (ATRX) helicase binds to clusters of G-quadruplex motifs, hence modulating the expression of these genes.40
Numerous studies on synthetic molecules that interact with G-quadruplexes have helped demonstrate the existence and elucidate putative biological roles of these nucleic acid structures.41–50 Previously reported quinoline-based molecules selectively recognize G-quadruplex structures over double-stranded DNA with high potency, making this chemical moiety an attractive feature to exploit for the design of G-quadruplex ligands.51–54 We previously reported a G-quadruplex stabilizing synthetic small molecule, based on a N,N′-bis(quinolinyl)pyridine-2,6-dicarboxamide scaffold (Fig. 1), which we named pyridostatin (1). We demonstrated the suitability of the molecule to target telomeres in cells.23 The design of these molecules was initially based on structural features shared by a potent G-quadruplex binding molecule54 with particular emphasis on (1) the ability to adopt a flat but flexible conformation, facilitated by an internal hydrogen bonding network, prone to adapt to the dynamic and polymorphic nature of diverse G-quadruplex structures; (2) an optimal electronic density of the aromatic surface to enable π–π interactions with the G-tetrad tuned by substituents (for instance alkoxy or halogens capable of altering the electron density) and (3) the presence of free nitrogen lone pairs able to coordinate with a molecule of water55 or alternatively to sequester a monovalent potassium cation in the centre, thus locking the flat surface of the molecule and facilitating the interaction with G-quartets (Fig. 1). These key features distinguish pyridostatin from other structurally related G-quadruplex interacting molecules.51–54 Prior to this work, we showed that pyridostatin, the lead compound of this family, induces telomere dysfunction by competing for binding with telomere associated proteins such as human POT1.23 Furthermore, we have illustrated the ability of a biotinylated analogue to mediate the selective pull-down of telomeric fragments from genomic DNA by means of affinity matrix isolation. During the course of this study, we have demonstrated the high selectivity of pyridostatin analogues towards G-quadruplex nucleic acids, regardless of sequence variability and structure polymorphism, compared to double-stranded DNA.56 Recent work in our laboratory has proven that pyridostatin alters transcription and replication of particular human genomic loci containing high G-quadruplex clustering within the coding region, which encompasses telomeres57 and selected genes such as the proto-oncogene SRC.58 These results emphasize that selective G-quadruplex ligands represent a new class of DNA damaging agents with limited sites of action throughout the genome, in contrast to well-known therapeutic agents previously reported to inducing genome-wide DNA damage in a stochastic manner associated with high cytotoxicity.
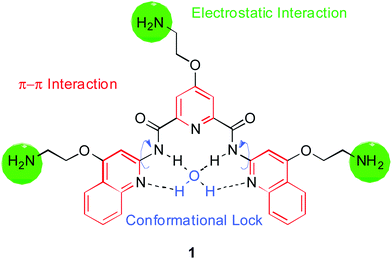 | ||
| Fig. 1 Molecular structure of pyridostatin (1). The design rationale is indicated on the structure. | ||
These results prompted us to synthesize structural variants based on the N,N′-bis(quinolinyl)pyridine-2,6-dicarboxamide scaffold to further explore the scope of their use as anti-cancer agents. We have used this family of compounds by keeping the central scaffold of pyridostatin intact and varying the substituents, including different cationic, neutral and glycosidic side-chains. These groups were systematically altered to gain further insights into structure–activity relationships. Herein, we describe an assessment of their potential to stabilize the telomeric G-quadruplex (H-Telo) and a selection of genomic promoter quadruplexes using a Förster Resonance Energy Transfer (FRET)-melting assay initially introduced by Mergny and Maurizot.59 Furthermore, we have measured their growth inhibitory properties against a panel of human cancer cell lines as a first potency readout, and finally have explored the phenotype induced at telomeres.
Results
Chemical synthesis of pyridostatin analogues
In this study, 26 small molecules based on the N,N′-bis(quinolinyl)pyridine-2,6-dicarboxamide scaffold were synthesized in two to seven synthetic steps as shown in Schemes 1 and 2. We chose to systematically vary side chain lengths and their chemical functionalities, keeping the central scaffold of pyridostatin unaltered. Synthetic analogues were obtained by coupling one mole equivalent of pyridine-based building blocks with two mole equivalents of quinoline-based building blocks as depicted in Scheme 1. The central core was synthesized either from chelidamic acid (2) or the commercially available pyridine-2,6-dicarbonyl dichloride (3). Compounds 4a and 4c were prepared by reacting 2 with thionyl chloride followed by quenching the reaction mixture with cold methanol (MeOH). The products were separated by aqueous work-up yielding 29% of 4a and 28% of 4c. Compound 4b was prepared in 90% yield from 2 in toluene–MeOH using (trimethylsilyl)diazomethane (Me3SiCHN2). Intermediate 4a was further functionalized via a Mitsunobu reaction, mixing diisopropyl azodicarboxylate (DIAD) and triphenylphosphine in tetrahydrofuran (THF) at 0 °C then room temperature. The resulting 4-substituted 2,6-pyridine-dimethylesters were then deprotected in the presence of sodium hydroxide (NaOH) in H2O–MeOH to afford the pyridine-based building blocks 5a–f in 57–77% yield. The quinoline-based building blocks were obtained in 53–71% yield from commercially available starting material 2-amino-quinolinone (6) either via a similar Mitsunobu reaction to provide 7a–e, or through a direct methylation in the presence of toluene–MeOH containing Me3SiCHN2. Building blocks 5a–f were reacted with Ghosez's reagent (1-chloro-N,N,2-trimethylpropenyl-amine) and then with triethylamine, followed by the addition of 7a–f to obtain products 1, 8–21 in 61–93% yield. Compounds 8–9, 11–12, 15–16 and 20 were then deprotected using trifluoroacetic acid (TFA) in dichloromethane (DCM). Compound 3 was reacted with 7a–g in the presence of triethylamine to provide compounds 22–26 in 62–82% yield. Finally, compounds 27–33 were synthesized in 22–67% yield from 11 and 12 using the copper catalyzed alkyne–azide 1,3-dipolar cycloaddition as shown in Scheme 2.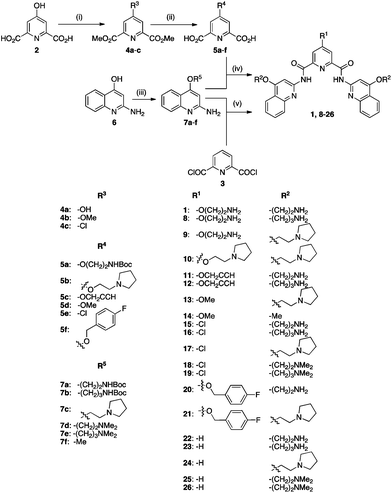 | ||
| Scheme 1 Synthetic route to compounds 1 and 8–26. (i) 4a, 4c: SOCl2, MeOH, 0 °C then rt, 2 h; 4b: toluene–MeOH, Me3SiCHN2, rt, 30 min; (ii) 5a–c, 5f: ROH, triphenylphosphine, DIAD, THF, 0 °C to rt, 3 d; then for 5a–f: NaOH (aq.), MeOH, rt, 1 h; (iii) 7a–e: ROH, triphenylphosphine, DIAD, THF, 0 °C to rt, 3 d; 7f: toluene–MeOH, Me3SiCHN2, rt, 30 min; (iv) 1, 8–21: 5a–f, 1-chloro-N,N,2-trimethylpropenyl-amine, DCM, 2 h, then triethylamine, 0 °C, 1 h then rt, then 7a–f, overnight; then for 1, 8, 9, 11, 12, 15, 16, 20: TFA–DCM, rt, 1 h; (v) 22–26: 2, triethylamine, DCM, 0 °C, 1 h, then 7a–f overnight; then for 22, 23: TFA–DCM, rt, 1 h. | ||
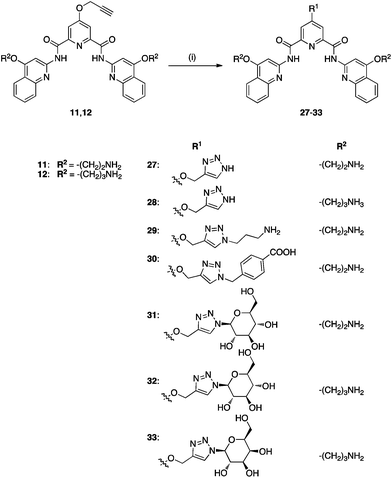 | ||
| Scheme 2 Synthetic route to compounds 27–33. (i) 27, 29–31: 11, CuSO4·5H2O, sodium L-ascorbate, RN3, H2O–tBuOH, rt, overnight; 28, 32, 33: 12, CuSO4·5H20, Sodium L-ascorbate, RN3, H2O–tBuOH, rt, overnight. | ||
Compounds 31–33 were conveniently obtained with retention of configuration, in mild conditions and without the use of protecting groups, highlighting the flexibility of this protocol.60 It is noteworthy that the condensed products of building blocks 5b, 5d and 5f with 7a–b, 7f could be precipitated from hot acetonitrile (MeCN) yielding either the final molecule, or a Boc (di-tert-butyl dicarbonate)-protected derivative, avoiding chromatographic purification, which then afforded the target molecule using standard TFA deprotection in DCM. Final products 1, 8–33 were further purified by high performance liquid chromatography (HPLC) to ensure high purity for the biophysical, in vitro and in vivo assays used in our subsequent studies. Molecules 22–26 all lack a side chain at position 4 of the pyridine ring, thus enabling us to investigate the relevance of that substitution for G-quadruplex stabilization and ensuing associated biological effects. Compounds 1, 8–14 carry side chains with varying chemical functionalities on the pyridine core as well as the quinoline moiety, which may influence the recognition towards different quadruplexes and relative conformations. We also introduced a chlorine onto the pyridine core (15–19) as well as a side chain containing a fluorinated benzene ring (20–21), which have the potential to participate in interactions with G-quadruplex loops or change the electron density of the pyridine core, and may thus provide additional selectivity over duplex DNA (ds-DNA).61 Compounds 27–33 were synthesized using the copper modified Huisgen reaction (Scheme 2), which enabled the introduction of complex functionalities such as sugar-containing asymmetric centers, capable of multiple hydrogen bond networking.
Biophysical evaluation of G-quadruplex stabilization by pyridostatin analogues
In order to study the interaction of the molecules with telomeric G-quadruplex DNA, we performed a Förster resonance energy transfer (FRET)-melting assay59 using the human telomeric G-quadruplex-forming sequence (H-Telo) and a ds-DNA as targets. The data are described in Table S1.† A large number of molecules of this family showed up to 35 K stabilization of H-Telo at 1 μM in melting experiments. None of the molecules studied showed any detectable stabilization of ds-DNA at this concentration, demonstrating the suitability of the scaffold for cell-based assays. Molecules 1, 8–10, which contain multiple amine functionalities, exhibited maximal stabilization for H-Telo at 1 μM ligand (Fig. 2, Table S1†).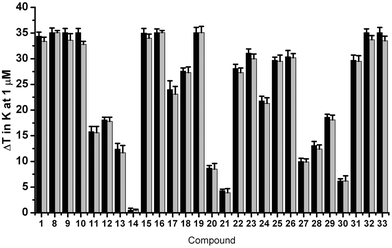 | ||
| Fig. 2 FRET-melting competition results at 1 μM for 1, 8–33 in the presence of 50 mol. equiv. of unlabeled ds-DNA against H-Telo. Values are expressed as ΔTm. Black: ΔTm in the absence of ds-DNA. Grey: ΔTm in the presence of ds-DNA. Errors denote the standard deviation of at least three independent experiments. | ||
Similarly, introducing other functions at position 4 of the pyridine moiety such as a chlorine (compounds 15–19) or sugars (compounds 31–33) improved the stabilization properties of the named compounds towards H-Telo compared to the unsubstituted analogues. This data set suggests that cation–dipole interactions as well as hydrogen and halogen-bonding interactions enable the improvement of the binding properties of the main scaffold towards G-quadruplex motifs. Conversely, removal of amine functionalities at positions R1 and R2, as for compounds 13–14, was found to reduce the stabilizing potential of the corresponding scaffold, thus further demonstrating the need for polar functionalities to promote G-quadruplex stabilization. Ligands carrying an alkyne functionality (11–12) induced moderate ΔTm-values, most likely due to the lack of the above mentioned stabilizing interactions. Ligands 27–29 also showed moderate stabilization of H-Telo, which was rather surprising for compound 29, since this analogue contains three positively charged functionalities that have been shown to be stability-enhancers for all the other members of this family. Additionally, the introduction of a negatively charged functionality to afford 30 hampered the ability of this analogue to interact with H-Telo, presumably due to electrostatic repulsion with the negatively charged phosphate backbone of the DNA. The 4-fluorobenzyloxy substitution at position R1 of ligands 20 and 21 resulted in lower ΔTm values of 8.8 K and 4.3 K, respectively. These results are consistent with those observed for alkyne substituted analogues and suggest that the steric hindrance imposed by the benzene and triazole rings at the central position may lead to a weaker contribution of the fluorine and amine substituents. However, this is not the case for compounds 31–33, since the sugar moieties might be involved in stabilizing non-covalent interactions, raising the ΔTm-values recorded for these compounds to above 30 K. The nature of the amine on the side chain at position R2 was found to have a small influence on the stabilization potential of the analogues following the general trend: –NMe2 ≥ –NH2 ≥ –pyrrolidine. Comparing ΔTm-values for 1 and 8, 15–16, 18–19, 22–23, 25–28 and 31–32, respectively, showed that a three-carbon side chain gives slightly higher stabilization potentials than two-carbon side chains. Finally, we have investigated the selectivity of each molecule for G-quadruplex over ds-DNA by measuring the melting of H-Telo in the presence of increasing amounts of each analogue and 50 mole equivalents of unlabelled ds-DNA competitor. Negligible changes in melting temperatures were observed, further demonstrating a high specificity of these compounds for quadruplex DNA over ds-DNA, as was shown before for this type of scaffold23,56 (see Fig. 2). Encouraged by these results, we next evaluated the stabilization properties of these compounds over various G4 structures before studying their aptitude to induce a phenotype in cell-based assays (see Fig. 3).
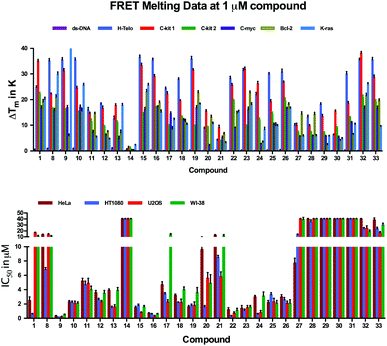 | ||
| Fig. 3 Representation of FRET melting values for various G-quadruplex structures (top) and IC50-values of growth inhibition after 72 h (bottom) treatment with compounds 1, 8–33. Errors denote the standard deviation of at least three independent experiments. | ||
Evaluation of cell viability upon exposure to analogues of pyridostatin
The molecules were assessed for their ability to inhibit cell growth using a luminescent cell viability assay. We investigated growth inhibition after 3 days of exposure to compounds 1, 8–33 on a panel of four human cell lines: HeLa (adenocarcinoma), HT1080 (fibrosarcoma), U2OS (osteosarcoma), and WI-38 (normal lung fibroblasts), the latter being non-cancerous. The data are summarized in Fig. 3 and Table S2.† Molecules 1, 9–10, 15–19 and 22–26 showed growth inhibition at high nanomolar to low micromolar concentrations against the panel of cell lines. Compound 9 displayed the lowest values of the series with IC50-values ranging from 0.2 to 0.5 μM. This suggests that positively charged side chains may be required for improving the water solubility and cellular uptake of the apolar central skeleton, in addition to participating in favorable non-covalent interactions with the DNA targets. It is noteworthy that most of the compounds showed generally lower IC50-values for the cancer cell lines than for the normal cell line WI-38. For example, pyridostatin (1) exhibited an 18.5-fold selectivity for HT1080 cells over WI-38 cells. Analogues 17 and 27 were found to be the most selective molecules for HeLa and U2OS with a 6.0- and 5.2-fold difference in growth inhibition over WI-38 cells, respectively. Notably, compounds 27–33, which contain a triazole at position R1, showed relatively higher IC50-values as compared to other analogues, which might reflect differences in cell permeability and uptake of these analogues, since the sugar functionalized compound retained remarkable stabilization properties. Ligand 14, which contains no functionalities, did not exhibit growth inhibitory properties at the concentrations used, thus reflecting on the poor solubility properties of this analogue and weak G-quadruplex stabilization potential. These results are in agreement with our previous reports23,58 showing that this family of small molecules promotes DNA double strand break formation, which results in cell cycle arrest and growth inhibition.Long-term growth inhibitory effects of pyridostatin analogues towards HT1080 cells
We investigated the long-term growth effects on HT1080 cells of a selection of molecules, namely 1, 9–10, 15–17, 19–24, 26–27 and 33, by incubating cells over a period of 30 days with the compounds (Fig. 4) at their respective IC50-values. Some of the molecules caused a strong reduction of population doublings indicating a long-term growth arrest of HT1080 cells, notably 1, 15 and 17 after 6 days, 9 after 12 days and 10 and 21 after 21 days of exposure. Compounds 20 and 33 also caused a decrease in population doublings over time, with a flattening of the curve starting after 19 and 25 days of exposure, respectively.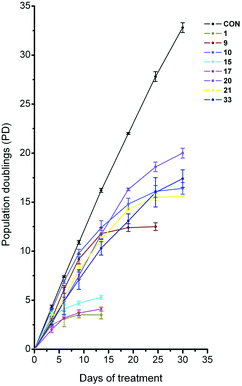 | ||
| Fig. 4 Studies of long-term growth effect of compounds 1, 9, 10, 15, 17, 20, 21 and 33 on HT1080 cells. CON denotes control cells, which were not treated with compound. Measurements were taken at the respective IC50 values of the compounds. | ||
Compounds 16, 19, 22–24, 26 and 27 did not show any additional decrease in population doublings over the time-course measured, as compared to what would be expected for incubation at the IC50. No change of population doublings was observed over the time course measured for compounds that did not have a substitution at position 4 of the pyridine ring (compounds 22–26). It is worth noting that 97–99% of the control and the cells treated at the respective IC50-value of each compound (1, 9–10, 15–17, 19–24, 26–27 and 33) were viable during the long-term treatment as observed by trypan blue nuclear exclusion, indicative of growth arrest as opposed to cytotoxicity induced by this family of compounds. In addition, the total cell number reached a plateau after long-term exposure without noticeable decrease, which also supports the absence of major cell killing. Our results indicate that complex mechanisms of action for these compounds are taking place. One of these mechanisms may involve the induction of dysfunctional telomeres, as previously shown for 1, due to a competition for binding of these analogues with telomere binding proteins and ensuing telomere shortening. To verify this hypothesis we investigated the influence of these analogues on telomere G-overhang integrity in HT1080 cells.
G-overhang shortening and cellular senescence induced by analogues of pyridostatin
Small molecules that can compete for binding with shelterin components have been shown to induce telomere shortening, G-overhang degradation and replicative senescence in cultured cells.23,47,56,62 We chose compounds 9–10, 15, 17 and 33, since these analogues displayed moderate to high stabilization of the telomeric G-quadruplex H-Telo as judged by FRET-melting assays and long-term growth arrest occurring between 6 to 25 days of treatment. A β-galactosidase assay performed on HT1080 cells after 8 days of incubation with compounds 9–10, 15, 17 and 33 at their respective IC50 revealed the presence of senescent cells (Fig. S4†). No senescent cells were observed in mock treated cells.We decided to measure telomere G-overhang lengths after treatment by employing a non-denaturing hybridization assay.62 The experiments were performed using a 21C oligonucleotide (5′-(CCCTAA)3CCC-3′) complementary to the 3′-G-overhang. G-overhang signals were assessed over a time course of 12 days, at five time-points (0 d, 3 d, 6 d, 9 d and 12 d). We found that all five analogues induced a decrease in the hybridization signal as compared to total genomic DNA stained with ethidium bromide (EtBr, Fig. 5). Consistent with a selective effect induced by the compounds, mock treated cells did not show any noticeable change in the hybridization signal. This showed that the telomeric G-overhang decreased in length over the time-course upon exposure to the small molecules. Interestingly, the kinetics of the G-overhang degradation appeared to vary, with compounds 10 and 17 being the most potent to induce an early effect, a result that is in apparent contrast with their kinetics of cell growth arrest (Fig. 4).
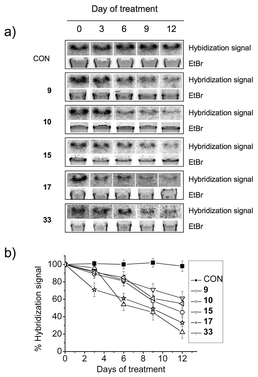 | ||
| Fig. 5 Telomeric G-overhang shortening assessed for 9, 10, 15, 17 and 33 in HT1080 cells by non-denaturing hybridization assay. CON represents DNA from cells grown in the absence of compound. Cells were incubated at the respective IC50 concentrations. (a) Hybridization gel radiograph and EtBr fluorescence picture. (b) Graph showing the %hybridization signal against days of treatment. The radiograph signal was normalized against the fluorescence signal. Errors denote the standard deviation from three different experiments. | ||
These data suggest that these compounds trigger a telomeric dysfunction associated with the induction of a phenotype reminiscent of cellular senescence. However, the correlation between telomere dysfunction and long-term growth arrest for compounds 9–10, 15, 17 and 33 is complex and suggests that additional events contribute to the growth inhibition and the induction of senescence.
Discussion
We have shown that N,N′-bis(quinolinyl)pyridine-2,6-dicarboxamides in general stabilize G-quadruplex nucleic acids selectively as compared to ds-DNA. Systematic changes of the side chains can help modulate their in vitro stabilization potential for G-quadruplexes and differentially affect short- and long-term cell growth. Moreover, their low molecular weight and global structural features confer drug-like properties. This constitutes a promising starting point for the development of compounds with differential growth inhibitory properties on different cell types combined with low toxicity.The synthesis of the library of compounds was efficient and gave moderate to high yields with high purity in 2–7 synthetic steps. In particular, the coupling of quinoline-based building blocks with the pyridine core using Ghosez’ reagent was facile and high yielding compared to other coupling strategies. We demonstrated that diverse simple and complex functional features could be introduced before or after the assembly of the main scaffold, which confers additional flexibility to our synthetic scheme. Functionalization via click chemistry, to yield molecules 27–33, was attractive for chain derivatization at a late stage of the synthesis.60 Molecules of this family have the ability to stabilize G-quadruplexes formed by the H-Telo sequence in the presence of an excess of ds-DNA (Fig. 2) and also a selection of promoter quadruplexes (Fig. S2 and Table S1†) with very high ΔTm values, demonstrating the versatility of this type of scaffold to bind G-quadruplex motifs with varying loop sequence. This unique property as a potent generic G-quadruplex binder rationalizes in part the ability of these compounds to target other genomic locations containing putative G-quadruplex forming motifs and to promote genomic instability at well defined genomic locations.23,58
The nature of the substitution at the pyridine moiety had a great influence on the stabilization potential of the molecules for H-Telo following the general trend of: amine > Cl > sugar > H > alkoxy. Positively charged functionalities and sugars conferred higher stabilization, most likely due to their potential for electrostatic and hydrogen bonding interactions with the nucleic acid target. Accordingly, substitution with uncharged, aromatic or aliphatic side chains generally had an adverse effect on the stabilization potential of the molecule, which could be due to electronic repulsion or steric clash. Modifications in the length of the side chain linked to the quinoline moiety had a less striking effect, but generally stabilization was higher for chain lengths of three carbons over two carbons. Notably, the stabilization potential for G-quadruplexes was almost eliminated for 14 with methoxy-substitutions on both R1 and R2. This shows that the side chains play a crucial role in the potential of the molecules to stabilize G-quadruplex nucleic acids. Clearly, the nature of side chains affected the water solubility properties of each analogue and may reflect differences observed in biophysical and biological assays.
Most of the molecules showed high nanomolar to low micromolar IC50-values for short-term (72 h) growth inhibition against the four cell lines investigated. Some of the compounds, in particular pyridostatin (1), 17, 24, and 27, showed a considerable difference in their growth-inhibitory properties between the different cell-lines investigated. Molecules 17, 24 and 27 were found to be more potent for cancer cell lines over WI-38. Compounds 17 and 24 are more selective towards the cancer cell lines than other analogues that carry a hydrogen or chlorine at position R1. This shows how the variation of the substitution at R2 can be used to modulate differential growth inhibitory properties from one cell line to another, which may be exploited further to develop small molecules empowered with selective anti-proliferative properties.
Long-term growth studies upon incubation with the compounds showed that some compounds caused a decrease in population doublings between 6 and 30 days treatment (Fig. 4). In this set of experiments each compound was screened at its IC50 value in order to detect and quantify a phenotype, thus enabling a comparative analysis of the analogues. The six compounds with the greatest differential effect on long-term growth were either substituted with an amine side chain (1, 9–10), a chlorine (15 and 17) or a sugar at position R1 (compound 33). Consistent with this, these compounds also exhibited very good stabilization of the telomeric quadruplex. None of the molecules substituted with a hydrogen at position R1 showed a considerable decrease in population doublings upon long-term exposure. Interestingly, all 6 analogues have either a primary amine or a pyrrolidine on the side chain at R2. The majority of the compounds that induced long-term growth arrest had a side-chain linked by two carbons (with the exception of compound 33). The cellular growth data suggests an important sensitivity to the R2 substitution. Compounds 16, 19 and 22–24 exhibit good short-term growth inhibition, rather than long-term growth effects, which suggest a distinction in biological mechanism(s). Potent stabilization of non-telomeric G-quadruplex structures by FRET-melting (Fig. S2 and Table S1†) may hint that non-telomeric G-quadruplex targeting is the predominant contributing factor to the observed short-term growth arrests. Indeed, our previous reports23,58 have shown that pyridostatin (1) induces DNA damage at non-telomeric loci of genomic DNA, resulting in the activation of a cell cycle checkpoint-dependent growth arrest.
Other G-quadruplex ligands have been shown to stabilize telomeric quadruplexes in human cells and cause telomere dysfunction and shortening leading to replicative senescence in telomerase-positive47 and alternative lengthening of telomeres (ALT)63,64 cells. Our investigation of the effect of some analogues (9–10, 15, 17 and 33) on the telomeric G-overhang revealed that these derivatives also trigger a telomere dysfunction in HT1080 cells, in agreement with our previous finding that pyridostatin competes for binding at telomeres with the G-overhang binding protein POT1 in HT1080 cells.23 However, the kinetics for long-term growth inhibition and G-overhang degradation largely differ between these compounds and suggest additional cellular mechanisms. Interestingly, all these derivatives induced senescence during long-term treatments (Fig. S4†) but did not induce an elevated level of cell death, suggesting a mechanism of action different from agents that cause a massive and global DNA damage response throughout the genome. This is in agreement with the targeting properties of N,N′-bis(quinolinyl)pyridine-2,6-dicarboxamides towards G-quadruplexes. Evidence indicates that telomere dysfunction is one of the causes underpinning cellular senescence.65 Therefore, the protracted accumulation of DNA damage at telomeres using low concentrations of chemotherapeutic agents to induce accelerated senescence is an increasingly known and investigated mechanism to inhibit tumor cell growth renewal.66 Thus, long-term growth arrests and senescence induced by these derivatives are noteworthy results of a complex interplay between telomeric effects and the accumulation of restricted DNA damage sites being generated throughout the genome via non-telomeric G-quadruplex targeting.
Conclusions
Small molecules with the ability to target telomeres are highly desirable for the treatment of malignant neoplasia, since one of the hallmark of cancer has been defined by the infinite proliferative capacity of cells permitted by mechanisms that sustain telomere length in addition to the ectopic expression of oncogenes.67 In general, most of the N,N′-bis(quinolinyl)pyridine-2,6-dicarboxamides that featured in our study showed excellent stabilization of the telomeric G-quadruplex combined with high selectivity over double-stranded DNA, making them suitable chemical agents to target telomeres. The compounds show some striking growth-inhibitory effects on cancer cell-lines after few days of exposure, some of them exhibiting a complete arrest after long-term exposure to the drug. The differential short and long-term inhibitory properties of the different analogues highlights the fact that subtle structural and functional chemical variations of the ligands can be exploited to fine-tune the biological activity of these analogues. Our data are in agreement with a model where both telomeric dysfunction and the accumulation of DNA damage sites at genomic loci enriched in G-quadruplex structures trigger a reduced cell growth associated with senescence induction. The selective targeting of G-quadruplexes by N,N′-bis(quinolinyl)pyridine-2,6-dicarboxamides constitute a promising angle for the development of novel anticancer drugs that could act by impairing cancer mechanisms such as telomere maintenance.Experimental
FRET-melting studies
100 μM stock solutions of oligonucleotides were prepared in molecular biology grade DNase-free water. Further dilutions were carried out in 60 mM potassium cacodylate buffer, pH 7.4. FRET experiments were carried out with a 200 nM oligonucleotide concentration. All labeled DNA oligonucleotides were supplied by Eurogentec® Ltd and all unlabeled oligonucleotides were supplied by IBA® GmbH. Seven dual fluorescently labeled DNA oligonucleotides were used in these experiments: H-Telo (5′-FAM-GGG TTA GGG TTA GGG TTA GGG-TAMRA-3′), C-kit1 (5′-FAM-GGG AGG GCG CTG GGA GGA GGG-TAMRA-3′), C-kit2 (5′-FAM-GGG CGG GCG CGA GGG AGG GG-TAMRA-3′), C-myc (5′-FAM-TGA GGG TGG GTA GGG TGG GTA A-TAMRA-3′), Bcl-2 (5′-FAM GGG CGC GGG ACG AGG GGG GCG GG-TAMRA-3′), KRAS (5′-FAM AGG GCG GTG TGG GAA GAG GGA AGA GGG GGA GG-TAMRA-3′), ds-DNA (5′-FAM-TAT AGC TAT A-HEG-T ATA GCT ATA-TAMRA-3′) which is a dual-labeled 20-mer oligonucleotide comprising a self-complementary sequence with a central polyethylene glycol linker able to fold into a hairpin. The competitor was an unlabeled 26-mer oligonucleotide (5′-CAA TCG GAT CGA ATT CGA TCC GAT TG-3′). The donor fluorophore was 6-carboxyfluorescein (FAM) and the acceptor fluorophore was 6-carboxytetramethylrhodamine (TAMRA). The dual-labeled oligonucleotides were annealed at a concentration of 400 nM by heating at 94 °C for 10 min followed by slow cooling to rt at a controlled rate of 0.1 °C min−1. 96-well plates were prepared by addition of 50 μl of the annealed DNA solution to each well, followed by 50 μl solution of molecules 1, 8–33 at the appropriate concentration. For the competition experiments, competitor was added to the fluorescently labeled quadruplex oligonucleotides sequences at an excess of 50 mol equiv. and annealed in the same solution. Measurements were made in triplicate with an excitation wavelength of 483 nm and a detection wavelength of 533 nm. Final analysis of the data was carried out using OriginPro 7.5 data analysis and graphing software (OriginLab®).Cell culture
HeLa, HT1080, U2OS and WI-38 cells were cultured in T-75 flasks in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal calf serum and split at 70–80% confluency using trypsin EDTA. The HT1080, WI-38 and U2OS cells were obtained from the European Collection of Cell Cultures (ECACC) and the HeLa cells were a generous gift of Prof. Ashok Venkitaraman/Cambridge.Luminescent cell viability assay
IC50 values of growth inhibition were determined using the cell viability assay CellTiter-Glo™ (Promega®). Cells were plated in 96 well plates at a density of 4000 cells per well in 100 μl of media and incubated for 24 h. Compounds 1, 8–33 were added in serial dilutions (in a range between 0–40 μM) at a volume of 100 μl per well at the respective concentrations. Cell viability was measured after 72 h using the manufacturer's protocol. Measurements were taken after 20 min incubation at rt. All measurements were made in triplicate. Final analysis of the data was carried out using OriginPro 7.5 data analysis and graphing software (OriginLab®).Long-term growth assay and cytotoxicity
HT1080 cells were cultured in T-25 flasks as stated above and seeded at 300000 cells per flask initially and incubated with 1, 9–10, 15–17, 19–24, 26–27 and 33 at their respective IC50 values or no compound as a control. The cells were split after 3 or 4 days, counted with a hemicytometer and reseeded at their initial density of 300000 cells per flask. This procedure was repeated every third day. All experiments were performed in triplicate. Population doubling (PD) was calculated using the following formula: PD = (ln(n1/n2)/ln2), n1 = initial number of cells, n2 = final number of cells. Cytotoxicity was assessed at each time point using a Trypan blue exclusion assay. Cells were counted (as stated above) in a 1:1 mixture of media:Trypan blue (0.4% in DMSO). Dead cells were assessed by blue nuclear staining.
β-Galactosidase assay
Senescence was assessed using a senescence β-galactosidase staining kit (Cell Signalling Technology®) according to the manufacturer's protocol. HT1080 cells were cultured in T-75 flasks for 1 d and incubated with compounds 9, 10, 15, 17 and 33 at their respective IC50 values for 4 d. As a control, cells were incubated without compound under the same conditions. Cells were split and seeded at 4000 cells per well on a Cover Well™ microscopy slide chamber (Invitrogen®) and incubated with compounds 9, 10, 15, 17 and 33 at the aforementioned concentrations or in the absence of compound for another 4 d. Subsequently, the media was removed and the wells washed twice with 1 × phosphate buffered saline (PBS). Cells were fixed with 2% formaldehyde and 0.2% glutaraldehyde in 1 × PBS for 15 min. This solution was then removed and the wells washed twice with 1 × PBS. Each well was incubated with 800 μl staining solution (40 mM citric acid/sodium phosphate (pH 6.0), 0.15 M NaCl, 2 mM MgCl2, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 1 mg ml−1 X-gal, 5% dimethylformamide (DMF)) at 37 °C for 16 h. The staining solution was removed and the cells stored in 70% glycerol.Non-denaturing hybridization assay
We performed a non-denaturing hybridization assay to detect the 3′ G-overhang. 5 μg of undigested genomic DNA, extracted from cells treated with 9, 10, 15, 17, 33 or no compound at the respective time points, were hybridized for 16 h at 50 °C with 0.5 pmol of [γ-32P] adenosine triphosphate (ATP)-labeled 21C oligonucleotide (5′-(CCCTAA)3CCC-3′) in 30 μl hybridization buffer (20 mM tris(hydroxymethyl)aminomethane (TRIS)·HCl (pH 8.0), 0.5 mM EDTA, 50 mM NaCl, 10 mM MgCl2). Reactions were stopped by the addition of 9 μl loading buffer (20% glycerol, 1 mM ethylenediaminetetraacetate (EDTA), 0.2% bromophenol blue) and size-fractioned on a 0.7% agarose gel for 3 h at 45 V. The gels were subsequently stained with ethidium bromide (EtBr). The bottom of the gel was cut to remove unbound probe and thus increase resolution. Gels were dried on Whatman® filter paper at 60 °C and exposed overnight on an autoradiography film. Ethidium bromide fluorescence and the autoradiography film were scanned with a phosphorimager (Typhoon™ 9210, Amersham Biosciences®). Results were expressed as the relative hybridization signal normalized to the fluorescent signal of EtBr.Alphabetical list of non-standard abbreviations
| ALT | alternative lengthening of telomeres |
| ATP | adenosine triphosphate |
| ATRX | alpha thalassemia/mental retardation syndrome X-linked |
| Boc | Di-tert-butyl dicarbonate |
| DIAD | diisopropyl azodicarboxylate |
| DCM | dichloromethane |
| DMEM | Dulbecco's modified Eagle medium |
| DMF | N,N-dimethylformamide |
| ds-DNA | double stranded DNA |
| EDTA | ethylenediaminetetraacetic acid |
| ESI | electrospray ionization |
| EtBr | ethidium bromide |
| FAM | 6-carboxyfluorescein |
| FC | flash chromatography |
| FRET | Förster resonance energy transfer |
| HPLC | high performance liquid chromatography |
| IC50 | half maximal inhibitory concentration |
| MeOH | methanol |
| NaOH | sodium hydroxide |
| NMR | nuclear magnetic resonance |
| PD | population doubling |
| PBS | phosphate buffered saline |
| POT1 | protection of telomeres 1 |
| TAMRA | 6-carboxytetramethylrhodamine |
| TERRA | telomeric repeat containing RNA |
| TFA | trifluoroacetic acid |
| THF | tetrahydrofuran |
| TEBP | telomeric end-binding protein |
| TLC | thin layer chromatography |
| T m | melting temperature |
| TRF1/2 | telometic repeat factor 1/2 |
| TRIS | tris(hydroxymethyl)aminomethane |
Acknowledgements
We thank Cancer Research UK for programme funding and for a studentship (S.M.), MUIR FIRB-Ideas RBID082ATK for programme funding (M.D.A.), Ligue Nationale Contre le Cancer for programme funding (J.F.R.), Helen Lightfoot for technical assistance, and Chris Lowe for carefully proofreading this manuscript.Notes and references
- I. Bang, Z. Physiol. Chem., 1901, 32, 201 CrossRef CAS.
- J. T. Davis, Angew. Chem., Int. Ed., 2004, 43, 668 CrossRef CAS.
- M. Gellert, M. N. Lipsett and D. R. Davies, Proc. Natl. Acad. Sci. U. S. A., 1962, 48, 2013 CrossRef CAS.
- S. B. Zimmerman, G. H. Cohen and D. R. Davies, J. Mol. Biol., 1975, 92, 181 CrossRef CAS.
- I. Cheung, M. Schertzer, A. Rose and P. M. Lansdorp, Nat. Genet., 2002, 31, 405 CAS.
- L. Cahoon and H. S. Seifert, Science, 2009, 325, 764 CrossRef CAS.
- P. Sarkies, C. Reams, L. J. Simpson and J. E. Sale, Mol. Cell, 2010, 40, 703 CrossRef CAS.
- K. Paeschke, J. A. Capra and V. A. Zakian, Cell, 2011, 145, 678 CrossRef CAS.
- K. Paeschke, T. Simonsson, J. Postberg, D. Rhodes and H. J. Lipps, Nat. Struct. Mol. Biol., 2005, 12, 847 CAS.
- R. K. Moyzis, J. M. Buckingham, L. S. Cram, M. Dani, L. Deaven, M. D. Jones, J. Meyne, R. L. Ratliff and J. R. Wu, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 6622 CrossRef CAS.
- G. N. Parkinson, M. P. H. Lee and S. Neidle, Nature, 2002, 417, 876 CrossRef CAS.
- Y. Wang and D. J. Patel, J. Mol. Biol., 1993, 234, 1171 CrossRef CAS.
- D. Sun, B. Thompson, B. E. Cathers, M. Salazar, S. M. Kerwin, J. O. Trent, T. C. Jenkins, S. Neidle and L. H. Hurley, J. Med. Chem., 1997, 40, 2113 CrossRef CAS.
- D. Gomez, T. Wenner, B. Brassart, C. Douarre, M.-F. O'Donohue, V. E. Khoury, K. Shin-ya, H. Morjani, C. Trentesaux and J.-F. Riou, J. Biol. Chem., 2006, 281, 38721 CrossRef CAS.
- A. Rizzo, E. Salvati, M. Purru, C. D'Angelo, M. F. Stevens, M. D'Incalci, C. Leonetti, E. Gilson, G. Zupi and A. Biroccio, Nucleic Acids Res., 2009, 37, 5353 CrossRef CAS.
- C. M. Azzalin, P. Reichenbach, L. Khoriauli, E. Giulotto and J. Lingner, Science, 2007, 318, 798 CrossRef CAS.
- A. M. Zahler, J. R. Williamson, T. R. Cech and D. M. Prescott, Nature, 1991, 350, 718 CrossRef CAS.
- D. J. Bearss, L. H. Hurley and D. D. V. Hoff, Oncogene, 2000, 19, 6632 CrossRef CAS.
- S. Neidle and G. Parkinson, Nat. Rev. Drug Discovery, 2002, 1, 383 CrossRef CAS.
- A. D. Cian, G. Cristofari, P. Reichebach, E. D. Lemos, D. Monchaud, M.-P. Teulade-Fichou, K. Shin-ya, L. Lacroix, J. Lingner and J.-L. Mergny, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17347 CrossRef.
- D. Gomez, A. Guédin, J. L. Mergny, B. Salles, J.-F. Riou, M.-P. Teulade-Fichou and P. Calsou, Nucleic Acids Res., 2010, 20, 7187 CrossRef.
- T. de Lange, Genes Dev., 2005, 19, 2100 CrossRef CAS.
- R. Rodriguez, S. Müller, J. A. Yeoman, C. Trentesaux and J.-F. Riou, J. Am. Chem. Soc., 2008, 130, 15758 CrossRef CAS.
- F. d'Adda di Fagagna, P. M. Reaper, L. Clay-Ferrace, H. Flegler, P. Carr, T. von Zglinicki, G. Saretzki, N. P. Carter and S. P. Jackson, Nature, 2005, 426, 194 CrossRef.
- J. L. Huppert and S. Balasubramanian, Nucleic Acids Res., 2005, 33, 2908 CrossRef CAS.
- J. L. Huppert and S. Balasubramanian, Nucleic Acids Res., 2007, 35, 406 CrossRef CAS.
- H. Fernando, A. P. Reszka, J. Huppert, S. Ladame, S. Rankin, A. R. Venkitaraman, S. Neidle and S. Balasubramanian, Biochemistry, 2006, 45, 7854 CrossRef CAS.
- S. Rankin, A. P. Reszka, J. Huppert, M. Zloh, G. N. Parkinson, A. K. Todd, S. Ladame, S. Balasubramanian and S. Neidle, J. Am. Chem. Soc., 2005, 127, 10584 CrossRef CAS.
- A. T. Phan, Y. S. Modi and D. J. Patel, J. Am. Chem. Soc., 2004, 126, 8710 CrossRef CAS.
- A. Siddiqui-Jain, C. Grand, D. J. Bearss and L. H. Hurley, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11593 CrossRef CAS.
- J. Dai, D. Chen, R. A. Jones, L. H. Hurley and D. Yang, Nucleic Acids Res., 2006, 34, 5133 CrossRef CAS.
- S. Cogoi and L. E. Xodo, Nucleic Acids Res., 2006, 34, 2536 CrossRef CAS.
- M. Bejugam, M. Gunaratnam, S. Müller, D. A. Sanders, S. Sewitz, J. A. Fletcher, S. Neidle and S. Balasubramanian, ACS Med. Chem. Lett., 2010, 1, 306 CrossRef CAS.
- M. Bejugam, S. Sewitz, P. S. Shirude, R. Rodriguez, R. Shahid and S. Balasubramanian, J. Am. Chem. Soc., 2007, 129, 12926 CrossRef CAS.
- S. Kumari, A. Bugaut, J. L. Huppert and S. Balasubramanian, Nat. Chem. Biol., 2007, 3, 218 CrossRef CAS.
- D. J. Patel, A. T. Phan and V. Kuryavyi, Nucleic Acids Res., 2007, 35, 7429 CrossRef CAS.
- C. Schaeffer, B. Bardoni, J.-L. Mandel and B. Ehresmann, EMBO J., 2001, 20, 4803 CrossRef CAS.
- A. Bugaut, R. Rodriguez, S. Kumari, S.-T. Hsu and S. Balasubramanian, Org. Biomol. Chem., 2010, 8, 2771 CAS.
- D. Gomez, T. Lemarteleur, L. Lacroix, P. Mailliet, J.-L. Mergny and J.-F. Riou, Nucleic Acids Res., 2004, 32, 371 CrossRef CAS.
- M. J. Law, K. M. Lower, H. P. J. Voon, J. R. Hughes, D. Garrick, V. Viprajasit, M. Mitson, M. D. Gobbi, M. Marra, A. Morris, A. Abbott, S. P. Wilder, S. Taylor, G. M. Santos, J. Cross, H. Ayyub, S. Jones, J. Ragoussis, D. Rhodes, I. Dunham, D. R. Higgs and J. R. Gibbons, Cell, 2010, 143, 367 CrossRef CAS.
- M. Di Antonio, F. Doria, S. N. Richter, C. Bertipaglia, M. Mella, C. Sissi, M. Palumbo and M. Freccero, J. Am. Chem. Soc., 2009, 131, 13132 CrossRef CAS.
- W. C. Drewe, R. Nanjunda, M. Gunaratnam, M. Beltran, G. N. Parkinson, A. P. Reszka, W. D. Wilson and S. Neidle, J. Med. Chem., 2008, 51, 7751 CrossRef CAS.
- K. Jantos, R. Rodriguez, S. Ladame, P. S. Shirude and S. Balasubramanian, J. Am. Chem. Soc., 2006, 128, 13662 CrossRef CAS.
- M. Y. Kim, H. Venkayalapati, K. Shin-Ya, K. Wierzba and L. H. Hurley, J. Am. Chem. Soc., 2002, 124, 2098 CrossRef CAS.
- C. Leonetti, S. Amodei, C. D'Angelo, A. Rizzo, B. Banassi, A. Antonelli, R. Elli, M. F. Stevens, M. D'Incalci, G. Zupi and A. Biroccio, Mol. Pharmacol., 2004, 66, 1138 CrossRef CAS.
- M. J. B. Moore, C. M. Schulters, J. Cuesta, F. Cuenca, M. Gunaratnam, F. A. Tanious, W. D. Wilson and S. Neidle, J. Med. Chem., 2006, 49, 582 CrossRef CAS.
- J.-F. Riou, L. Guittat, P. Mailliet, A. Laoui, E. Renou, O. Petitgenet, F. Mégnin-Chanet, C. Hélène and J.-L. Mergny, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2672 CrossRef CAS.
- R. Rodriguez, G. D. Pantoş, D. P. N. Gonçalves, J. K. M. Sanders and S. Balasubramanian, Angew. Chem., Int. Ed., 2007, 46, 5404 Search PubMed.
- J.-H. Tan, T.-M. Ou, J.-Q. Hou, Y.-J. Lu, S.-L. Huang, H.-B. Luo, J.-Y. Wu, Z.-S. Huang, K.-Y. Wong and L.-Q. Gu, J. Med. Chem., 2009, 52, 2825 CrossRef CAS.
- Z. A. E. Waller, P. S. Shirude, R. Rodriguez and S. Balasubramanian, Chem. Commun., 2008, 1467 RSC.
- A. D. Cian, E. DeLemos, J.-L. Mergny, M.-P. Teulade-Fichou and D. Monchaud, J. Am. Chem. Soc., 2007, 129, 1856 CrossRef.
- C. Granotier, G. Pennarun, L. Riou, F. Hoffschir, L. R. Gauthier, A. D. Cian, D. Gomez, E. Mandine, J.-F. Riou, J.-L. Mergny, P. Mailliet, B. Dutrillaux and F. D. Boussin, Nucleic Acids Res., 2005, 33, 4182 CrossRef CAS.
- S. Müller, G. D. Pantoş, R. Rodriguez and S. Balasubramanian, Chem. Commun., 2009, 80 RSC.
- P. S. Shirude, E. R. Gillies, S. Ladame, F. Godde, K. Shin-Ya, I. Huc and S. Balasubramanian, J. Am. Chem. Soc., 2007, 129, 11890 CrossRef CAS.
- S. L. Jain, P. Bhattacharyya, H. L. Milton, A. M. Z. Slawin, J. A. Crayston and J. D. Woollins, Dalton Trans., 2004, 862 RSC.
- S. Müller, S. Kumari, R. Rodriguez and S. Balasubramanian, Nat. Chem., 2010, 2, 1095 CrossRef.
- D. Koirala, S. Dhakal, B. Ashbridge, Y. Sannohe, R. Rodriguez, H. Sugiyama, S. Balasubramanian and H. Mao, Nat. Chem., 2011, 3, 782 CrossRef CAS.
- R. Rodriguez, K. M. Miller, J. V. Forment, C. R. Bradshaw, M. Nikan, S. Britton, T. Oelschlaegel, B. Xhemalce, S. Balasubramanian and S. P. Jackson, Nat. Chem. Biol., 2011, 8, 301 CrossRef.
- J.-L. Mergny and J.-C. Maurizot, ChemBioChem, 2001, 2, 124 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 48, 16789 CrossRef.
- D. Gomez, R. Paterski, T. Lemarteleur, K. Shin-Ya, J. L. Mergny and J.-F. Riou, J. Biol. Chem., 2004, 279, 41487 CrossRef CAS.
- A. J. Cesare and R. R. Reddel, Nat. Rev. Genet., 2010, 11, 319 CrossRef CAS.
- N. Temime-Smaali, L. Guittat, A. Sidibe, K. Shin-ya, C. Trentesaux and J.-F. Riou, PLoS One, 2009, 4, 6919 Search PubMed.
- F. d'Adda di Fagagna, Nat. Rev. Cancer, 2008, 8, 512 CrossRef CAS.
- D. A. Gewirtz, S. E. Holt and L. W. Elmore, Biochem. Pharmacol., 2008, 76, 947 CrossRef CAS.
- D. Hanahan and R. Weinberg, Cell, 2011, 144, 646 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization of building blocks and pyridostatin analogues and FRET-melting profiles. See DOI: 10.1039/c2ob25830g |
| This journal is © The Royal Society of Chemistry 2012 |