Discovery, application and protein engineering of Baeyer–Villiger monooxygenases for organic synthesis
Kathleen
Balke
,
Maria
Kadow
,
Hendrik
Mallin
,
Stefan
Saß
and
Uwe T.
Bornscheuer†
*
Institute of Biochemistry, Dept of Biotechnology & Enzyme Catalysis, Greifswald University, Felix-Hausdorff-Str. 4, 17487 Greifswald, Germany. E-mail: uwe.bornscheuer@uni-greifswald.de; Fax: +49 3834 8679 4367; Tel: +49 3834 864367
First published on 13th June 2012
Abstract
Baeyer–Villiger monooxygenases (BVMOs) are useful enzymes for organic synthesis as they enable the direct and highly regio- and stereoselective oxidation of ketones to esters or lactones simply with molecular oxygen. This contribution covers novel concepts such as searching in protein sequence databases using distinct motifs to discover new Baeyer–Villiger monooxygenases as well as high-throughput assays to facilitate protein engineering in order to improve BVMOs with respect to substrate range, enantioselectivity, thermostability and other properties. Recent examples for the application of BVMOs in synthetic organic synthesis illustrate the broad potential of these biocatalysts. Furthermore, methods to facilitate the more efficient use of BVMOs in organic synthesis by applying e.g. improved cofactor regeneration, substrate feed and in situ product removal or immobilization are covered in this perspective.
 Kathleen Balke | Kathleen Balke (born 1986) studied biochemistry at the University of Greifswald. During her studies she performed research internships at Dr Reddy's Chirotech Technology in Cambridge, UK and in the group of A. Achour at the Karolinska Institute in Stockholm, Sweden. She finished her diploma thesis in 2012 in the group of Prof. Bornscheuer on Baeyer–Villiger monooxygenases. |
 Maria Kadow | Maria Kadow (born 1984) studied biochemistry at the University of Greifswald and obtained her diploma degree in 2009. Since then she performs her PhD studies in the group of Uwe Bornscheuer. Her research topics deal with the identification of novel Baeyer–Villiger monooxygenases and the synthetic application of these enzymes. |
 Hendrik Mallin | Hendrik Mallin (born 1985) studied biochemistry in Greifswald, Germany. During his diploma thesis in the Bornscheuer group he worked on protein engineering of oxidative enzymes. In 2010 he started his PhD under supervision of Uwe Bornscheuer in which he investigates process development for organic synthesis with oxidative enzymes and transaminases. His research interests include immobilization of proteins, protein engineering and plasma techniques for process improvement. |
 Stefan Saß | Stefan Saß (born 1982) studied biochemistry at the University of Greifswald and obtained his diploma degree in 2009. His PhD thesis under supervision of Uwe Bornscheuer is focused on the establishment of screening systems for Baeyer–Villiger monooxygenases and the application of protein engineering. |
 Uwe T. Bornscheuer | Uwe T. Bornscheuer (born 1964) studied chemistry and completed his doctorate in 1993 at the University of Hannover. He then was a postdoc at the University of Nagoya (Japan). In 1998, he completed his Habilitation at the University of Stuttgart and was appointed Professor at Greifswald University in 1999. Bornscheuer edited and wrote several books and is Co-Chairman of the journal ChemCatChem. In 2008, he received the Biocat2008 Award for his innovative work in biocatalysis and in 2012 the Chevreul Medal for his pioneering work in enzymatic lipid research. His current research interest is focused on protein engineering of enzymes from various classes with special emphasis on applications in organic synthesis. |
Introduction
Baeyer–Villiger monooxygenases (BVMOs) catalyze the enzymatic counterpart of the chemical Baeyer–Villiger oxidation and both are important for synthetic organic chemistry. In contrast to standard chemical oxidants such as peracids or hydrogen peroxide, BVMOs offer the unique advantage that they show usually excellent regio- and stereoselectivity and hence provide an easy and mild method to obtain optically and regioisomerically pure products. Furthermore, the use of protecting groups and formation of by-products can be avoided in enzymatic processes.The occurrence and properties of natural and recombinant enzymes, and the broad synthetic utility of BVMOs have been reviewed in the past few years.1–5 This article will concentrate therefore on two aspects: (i) the recent advances in discovery and protein engineering of BVMOs to broaden their synthetic utility and (ii) new applications in organic synthesis, optimized reaction systems and immobilization methods to enable the efficient use of BVMOs in biotransformation.
Discovery and recombinant expression
Until the mid-1990s, research with Baeyer–Villiger monooxygenases was mostly restricted to two microorganisms, Acinetobacter calcoaceticus6 and Pseudomonas putida.7A. calcoaceticus produces a BVMO with high activity in the conversion of cyclohexanone to ε-caprolactone and hence the enzyme is usually named cyclohexanone monooxygenase (CHMOAcineto). The limitation that this strain is pathogenic and can only be grown in laboratories with proper permission (L2) was overcome by Stewart et al. as they succeeded first in the cloning and functional expression of this CHMO8,9 paving the way for easier studies of this enzyme. The Pseudomonas putida strain NCIMB 10007 was shown to contain three BVMOs7,10–14 but until recently (see below), the enzymes could only be used as purified proteins isolated after cultivation of the strain.Novel BVMOs from prokaryotic origin
In the past few years a tremendous increase in the number of new BVMOs took place due to the fast-growing information deposited in public sequence databases, which in combination with BVMO-specific amino acid motifs led to the identification, cloning, expression and characterization of numerous enzymes. Almost all recombinantly available BVMOs belong to the class of type I BVMOs. Type I BVMOs are NADPH and FAD dependent. Type II BVMOs that are FMN and NADH dependent have not been investigated in detail until recently. Type I BVMOs contain some typical motifs – such as FXGXXXHXXXW[P/D] described in 2002 by Fraaije et al.15 – that facilitate identification of putative BVMOs from sequence data and are therefore called fingerprint motifs. Another conserved motif in type I BVMOs is the N-terminal GXGXXG Rossmann-fold motif of which two enclose the fingerprint. One example of an enzyme that has been identified by genome mining using the fingerprint motif is the phenylacetone monooxygenase (PAMO) from Thermobifida fusca.16 Until now PAMO is the only available thermophilic BVMO and it was the first type I BVMO of which the structure was determined.17 The most impressive example of newly identified BVMOs is the discovery of over 20 putative BMVOs found in the genome of Rhodococcus jostii RHA1.18,19 Even though BVMOs are present in a variety of bacteria and fungi, usually only a few BVMOs are encoded in the genome of one specific strain and hence Rhodococcus jostii RHA1 is exceptional. Recently Riebel et al. succeeded in cloning and expressing 22 BVMOs from this strain.18 Additionally, 39 substrates were tested with each of these BVMOs in order to explore their substrate scopes. In earlier studies the investigation of those BVMOs was incomplete due to problems expressing some of the BVMOs.19 In comparison to Szolkowy et al., the Fraaije group was also able to identify one additional BVMO (BVMO24) in the proteome by comparing the protein sequences of PAMO and CHMO with potential BVMOs and completed the gene of another BVMO that had been suggested to lack a large part of the C-terminus (BVMO8). One of the earlier identified BVMOs was discarded from the recent study since it was found to be an FMO (BVMO22; FMOs are human flavin-containing monooxygenases) and BVMO23 was excluded because it only differed in one amino acid from BVMO21. By comparing the sequences of the thus obtained 22 BVMOs, the typical BVMO motifs mentioned above and slightly mutated forms of the fingerprint motif were found in all these BVMOs. Additionally, another motif, which is located between the N-terminal and the BVMO motif, was identified ([A/G]GXWXXXX[F/Y]P[G/M]XXXD). This motif was supposed to be more suitable for identifying new BVMOs because it contains more conserved residues and allows differentiation between BVMOs and FMOs. Of the 22 investigated BVMOs eight did not show any activity to the substrates tested and five BVMOs converted ten or more substances. Two of these BVMOs (BVMO4 and BVMO24) seemed to be very potent biocatalysts since they accepted a large number of substrates. Their substrate scope was similar to that of cyclopentanone monooxygenase from Comamonas testosterioni NCIMB 9872 (CPMOComa). For BVMO9 and 15, the substrate scope was shown to be similar to 4-hydroxyacetophenone monooxygenase from Pseudomonas fluorescens ACB (HAPMOACB) as they converted mostly aromatic ketones. In addition to a spectrophotometric assay used for these studies, some of the BVMOs were also analyzed with typical BVMO substrates including prochiral sulfides by GC analyses. Phenylacetone and bicyclo[3.2.0]hept-2-en-6-one were converted by all BVMOs even though conversions differed somewhat. BVMO8 showed the lowest conversions and BVMO24 differed from the other enzymes with respect to preferred substrate and extent of conversion, and it showed opposite enantiopreference towards the prochiral sulfides. Hence this study alone substantially extended the number of characterized BVMOs.A very promising representative of newly available BVMOs is the cyclopentadodecanone monooxygenase (CPDMO) from Pseudomonas HI-70. This enzyme was already isolated in 200620 when its low protein sequence similarity to the enzymes known at that time was ascertained. While highest catalytic efficiency of this BVMO was detected for cyclopentadecanone, good activity towards large ring ketones (C11–C13) and substituted cyclohexanones was also shown. Later, the enzymes’ activity and high selectivity on ketosteroids was confirmed.21 Recently, extensive profiling of the substrate scope of CPDMO and its revisited integration in the phylogenetic relationship of currently known BVMOs yielded interesting new features of this enzyme.22 From a present day perspective, CPDMO belongs to a newly identified branch of BVMOs, which was then named after this specific enzyme. Cycloketone- and arylketone-converting enzymes can be found in the vicinity of the CPDMO-branch, whereas these enzymes appear to be separated from the CHMO- and CPMO-clusters. Interestingly, another class of newly identified enzymes, the 1-deoxy-11-oxopentalenic acid monooxygenases, which will later be discussed in detail, also belongs to the CPDMO-branch. For an actual example of a comprehensive phylogenetic tree, we refer to the article by Leipold et al. where a newly discovered cycloalkanone monooxygenase from eukaryotic origin is described.23 CPDMO was shown to oxidize a variety of substituted cyclobutanones and -hexanones as well as fused and bridged bi- and tricyclic ketones. These results were compared to the best-known candidates for the respective compound.22 Within desymmetrization reactions a similar substrate scope and identical stereopreference compared to known members of the CHMO-cluster were detected. While conversion and enantioselectivity in general did not exceed other BVMOs like CHMO from Xanthobacter sp. ZL5 (CHMOXantho),24 improved performance concerning sterically demanding substituted cyclohexanones was observed. This however did not pertain to 4-methyl-4-phenyl substituted cyclohexanone, wherefore it was assumed that the ability of CPDMO to oxidize large compounds is not a general feature of this enzyme, but rather restricted to particular substrates.
There are only a few type II BVMOs known and two of them are involved in the camphor degradation pathway of Pseudomonas putida ATCC 17453 (identical to NCIMB 10007). These BVMOs were named 2,5-diketocamphane-1,5-monooxygenase (2,5-DKCMO) and 3,6-diketocamphane-1,6-monooxygenase (3,6-DKCMO) and are responsible for the conversion of the two isomers of diketocamphane that are formed through the degradation of (+)- and (−)-camphor.10,25 The other known type II BVMOs are two FMN and NADH dependent luciferases from Photobacterium phosphoreum NCIMB 844 and from Vibrio fischeri ATCC 7744 for which a Baeyer–Villiger oxidation of 2-tridecanone and some mono- and bicyclo[3.2.0]ketones was observed.26 Moreover, a type II BVMO being involved in the degradation of limonene in Rhodococcus erythropolis has been described.27 Type II BVMOs are of special interest for industrial application since they depend on the cofactor NADH, which is much cheaper than NADPH and therefore the recent identification of the genes encoding the type II BVMOs in the camphor degradation pathway, their recombinant expression and characterization has been a gain for biocatalysis.12,13 It was shown that the DKCMOs mainly convert bicyclic ketones such as camphor and (±)-cis-bicyclo[3.2.0]hept-2-en-6-one, but they are also able to convert monocyclic ketones and α,β-unsaturated monocyclic ketones (Table 1).
| Substrate | Conv.a (%) | Conv.b (%) |
|---|---|---|
| a With 2,5-DKCMO. b With 3,6-DKCMO; n.d. = not determined. | ||
| (+)-Camphor | 66 | 88 |
| (−)-Camphor | 25 | 91 |
| Cyclobutanone | n.d. | 13 |
| Cyclopentanone | n.d. | 24 |
| Cyclohexanone | n.d. | 3 |
| Acetophenone | n.d. | 80 |
| 4-Phenyl-2-butanone | n.d. | 48 |
| 2-Decanone | n.d. | 11 |
| Norcamphor | 98 | 77 |
| (±)-cis-Bicyclo[3.2.0]hept-2-en-6-one | 100 | 99 |
| (R,R)-Bicyclo[2.2.1]heptanes-2,5-dione | 94 | 26 |
| 2-Cyclopenten-1-one | 38 | 48 |
| 3-Methyl-2-cyclopenten-1-one | 11 | 10 |
| 2,3,4,5-Tetramethyl-2-cyclopenten-1-one | 44 | 43 |
| 2-Cyclohexen-1-one | 58 | 50 |
| 3-Methyl-2-cyclohexen-1-one | 19 | 20 |
| 3,5,5-Trimethyl-2-cyclohexen-1-one | 27 | 21 |
However, the great limitation in the efficient application of the DKCMOs is their additional need for a suitable reductase. In contrast to type I BVMOs, where oxygenating and flavin reducing subunits are combined in one polypeptide chain, type II BVMOs need a separate FMN reductase for regeneration of the cofactor. Until now, it is not known how the reduced flavin reaches the monooxygenase active site and if the reductase is bound to the monooxygenase throughout the reaction. Type II BVMOs lack the typical structural features of type I BVMOs such as the fingerprint domain and the GXGXXG-motifs.
The only available structure of a type II BVMO has been determined for 3,6-DKCMO,28 while there are a few known structures of type I BVMOs. Recently the structure of 2-oxo-Δ3-4,5,5-trimethylcyclopentenylacetyl-CoA-monooxygenase (OTEMO) has been published.11 This enzyme is the third BVMO involved in the degradation of camphor to isobutyrate. Its natural function seems to be the conversion of a cyclopentenylacetyl-CoA derivative, leading to the assumption that it is a suitable biocatalyst for monocyclic ketones. Two separate studies have investigated the biochemical properties and substrate specificity of recombinant OTEMO.11,12 Kadow et al. found that OTEMO prefers bicyclic ketones over monocyclic ketones and that it is a good catalyst for unsaturated cycloketones (Table 2). It was proposed that this is due to the fact that the natural substrate of OTEMO is a CoA-derivative and that possibly the corresponding monocyclic-CoA derivatives would be better accepted. In the work by Leisch et al. it was then shown that OTEMO indeed exhibits the highest affinity to the CoA-activated 2-oxo-Δ3-4,5,5-trimethylcyclopentenylacetic acid (KM 18 μM) and also converts the CoA-derivative with a higher rate (kcat 4 s−1) than the free acid (kcat 0.13 s−1).11 Kinetic parameters were determined for several other substrates as well (Table 2). Additionally, kinetic resolutions of racemic ketones have been performed, revealing a high enantioselectivity (E-value) of OTEMO towards 2-methylcyclopentanone (E > 200) in comparison to CHMO from Rhodococcus sp. HI-31 (CHMORhodo), where an E-value of only 1.4 was determined. On the other hand E-values for cyclohexanone-derivatives were much lower than the ones determined for CHMORhodo. Desymmetrization reactions with prochiral 4-substituted cyclohexanones showed that OTEMO provides enantiocomplementarity behavior to the CHMORhodo. Thus, both antipodes of the lactones derived from these prochiral ketones are available when using OTEMO or CHMORhodo. In contrast to CHMORhodo and PAMO, OTEMO functions as a dimer. The monomer structure of OTEMO, however, is closely related to the structures of CHMORhodo and PAMO. The published structure of OTEMO (pdb-code: 3UP5) is the first dimeric structure of a BVMO with bound cofactors.
| Substrate | Conv. (%) | k cat/KM (s−1 mM−1) |
|---|---|---|
| n.d. = not determined, OTE-CoA: 2-oxo-Δ3-4,5,5-trimethylcyclopentenylacetyl-CoA. | ||
| (+)-Camphor | 44 | n.d. |
| (−)-Camphor | 22 | n.d. |
| Cyclobutanone | 36 | 14.7 |
| Cyclopentanone | 19 | n.d. |
| Cyclohexanone | 19 | n.d. |
| Acetophenone | 67 | n.d. |
| 4-Phenyl-2-butanone | 54 | n.d. |
| 2-Decanone | 7 | n.d. |
| Norcamphor | 96 | n.d. |
| (±)-cis-Bicyclo[3.2.0]hept-2-en-6-one | 100 | 49.3 |
| (R,R)-Bicyclo[2.2.1]heptanes-2,5-dione | 87 | n.d. |
| 2-Cyclopenten-1-one | 62 | n.d. |
| 3-Methyl-2-cyclopenten-1-one | 13 | n.d. |
| 2,3,4,5-Tetramethyl-2-cyclopenten-1-one | 34 | n.d. |
| 2-Cyclohexen-1-one | 74 | n.d. |
| 3-Methyl-2-cyclohexen-1-one | 13 | n.d. |
| 3,5,5-Trimethyl-2-cyclohexen-1-one | 22 | n.d. |
| OTE-CoA | n.d. | 270 |
| 2-Oxocyclopentylethylacetate | n.d. | 22 |
| 2-Oxocyclohexylethylacetate | n.d. | 5.6 |
| 2-Methylcyclohexanone | n.d. | 4 |
| 4-Methylcyclohexanone | n.d. | 3 |
| 2-n-Hexylcyclopentanone | n.d. | 430 |
The first report on fungal Baeyer–Villiger oxidation of steroids over 50 years ago discussed the conversion of progesterone to testololactone in Penicillium species and Aspergillus flavus.29 In an actual study, focus was given on the capability of producing steroidal lactones by strains outside the genera Penicillium and Aspergillus.30 The soil fungus Beauveria bassiana KCH 1065 was chosen because differences in its metabolic pathway of dehydroepiandrosterone (DHEA), androstenedione and progesterone were reported in the literature.31,32 BVMOs acting on steroids (steroid monooxygenases) in general exhibit a rather narrow substrate spectrum, as they are able to catalyze oxidation of steroidal substrates only. Thereby the most common reaction is the oxidation of the C-17 and/or C-20 carbonyl group in 4-en-3-oxo steroids. The BVMO-activity of B. bassiana is distinguished from those enzymes by the fact that it oxidizes solely substrates with an 11α-hydroxyl group. The presence of the D-lactone without the 11α-hydroxyl group was not detected. Although this approach provides interesting new insights on BVMO activity in fungal steroid metabolism, the identification of the responsible enzymes and their cloning is strongly awaited as it would render experimental proof on the distinct role of the enzyme in the pathway and this might open a large field of new applications keeping in mind that steroid lactones provide anticancer, antiandrogenic, and antihypercholesterolemic properties.
Novel BVMOs from eukaryotic origin
Until 2011 all recombinantly produced type I BVMOs have been of prokaryotic origin. The first BVMO from a eukaryotic organism to be cloned and expressed was the cycloalkanone monooxygenase (CAMO) from the ascomycete Cylindrocarpon radicicola ATCC 11011.23 This strain, also known as Ilyonetria radicicola DSM 837, was reported to convert progesterone via androstenedione towards Δ1-dehydrotestololactone.29 Those Baeyer–Villiger-reactions were supposed to be catalyzed by only one enzyme.33 Additionally, C. radicicola was known to convert bicyclic ketones representing typical CHMO substrates as well.34 Leipold et al. recently succeeded in identifying a BVMO gene in this strain by CODEHOP PCR. This BVMO showed 46.4% sequence identity to the CHMO from Rhodococcus sp. Phil35 and 44.1% to CHMOAcineto36 and was thus claimed to be a CHMO-like BVMO. However, this newly identified BVMO differs from typical known CHMOs. Firstly, the consensus motif in this BVMO is FXGXXXHXXXWD and not FXGXXXHXXXWP as was found for certain type I BVMOs like CHMOs and PAMO. Also the temperature at which the enzyme retains half of its initial activity determined for CAMO is with 26 °C significantly lower than for CHMOAcineto with 36 °C. Leipold et al. suggested that this might be due to a methionine residue (Met57) in close proximity to the reactive C4 atom from FAD, which is not present in CHMOs or PAMO. Additionally, two of the ten residues forming the binding pocket of CAMO differ from the ones found in CHMOs, where all of these residues are conserved. In CAMO, residues 435 and 437 are alanine and phenylalanine, whereas in CHMOs those residues are threonine and leucine, respectively. Since the residues present in CAMO are smaller, leading to a larger binding pocket, the acceptance of a broad range of substrates might be explained. CAMO was shown to convert cycloaliphatic ketones, open-chain ketones and bicyclic ketones. Indeed, the highest kcat/KM value was observed for cyclobutanone. However, CAMO did not convert any of the tested steroids, which means that this enzyme is not responsible for the conversion of progesterone, indicating that there are several BVMOs encoded in the genome of C. radicicola.BVMO-activity of flavin-containing monooxygenases
During the previous years it was thought that BVMOs were fully absent in the genomes of archea, plants and higher organisms, but recently the performance of Baeyer–Villiger reactions by human enzymes was observed.37 In that study, the oxidation of a 4-hydroxypiperidine moiety by human flavin-containing monooxygenase 5 (FMO5) in a Baeyer–Villiger reaction was observed. The BVMO-substrate 10-((4-hydroxypiperidin-1-yl)methyl)chromeno[4,3,2-de]phthalazin-3(2H)-one (1, Scheme 1) is a potential anticancer agent because it acts as an inhibitor of poly(ADP-ribose) polymerase. The apparent oxidation and ring opening of this compound has been observed during preclinical studies on animals. In humans, five FMO isoforms are present that show a tissue-specific distribution while FMO5 occurs in adult human liver and small intestine. Their biological role is the detoxification of drugs and other xenobiotics into more hydrophilic metabolites. Typical FMO-catalyzed reactions are the monooxygenation of heteroatoms such as nitrogen, sulfur, and phosphorus, but the Baeyer–Villiger oxidation of salicylaldehyde to pyrocatechol by human FMO1 and the existence of an almost identical sequence motif in the active sites of FMOs and BVMOs have been shown as well.15 Due to the identification of the ring-opened hydroxyl carboxylic acid in incubations of hepatocytes from different species, it was assumed that these cells provide the necessary enzymes to first transform the 4-hydroxypiperidine into a ketone by an oxidoreductase and then oxidize this intermediate via a Baeyer–Villiger reaction to lactone 2 in liver microsomes. The lactone could afterwards be hydrolyzed to produce the ring-opened acid. This hypothesis was confirmed by investigations using recombinant enzymes.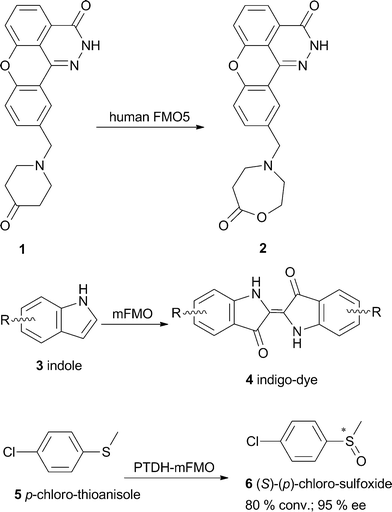 | ||
| Scheme 1 Substrate scope of human flavin monooxygenases and mFMO from Methylophaga sp. | ||
Inspired by the close homology between FMOs and BVMOs, different typical BVMO-substrates like 2-octanone, cyclohexanone and acetophenone have recently been subjected to mFMO from Methylophaga sp. strain SK1.38 This enzyme aroused researchers’ interest because it originates from bacteria and is therefore soluble in contrast to human FMOs, which are often membrane-bound. Although no activity towards the substrates mentioned could be detected, the oxidation of indole 3 and analogues into the corresponding indigoid pigment 4, which represent interesting dyes, was observed (Scheme 1). Moreover, enzymatic sulfoxidation of prochiral sulfides like p-chlorothioanisole 5 with excellent enantioselectivity was observed. Although FMOs have only rarely been shown to catalyze typical Baeyer–Villiger oxygenations, their potential use in biotransformation appears interesting due to their dependency on NADH as a cofactor.
Very recently, the BV-oxidation of bicyclo[3.2.0]hept-2-en-6-one by the flavin-containing monooxygenase from Stenotrophomonas maltophilia (SMFMO) was described.39 The 38.6 kDa FAD-containing protein was shown to favor NADH over NADPH as a cofactor and to catalyze the conversion of prochiral aromatic thioethers like p-chlorophenyl methyl sulfide with 80% ee of the (R)-product. Furthermore, the 3D-structure of SMFMO (Uniprot B2FLR2) was reported in this work. Within FMOs and BVMOs with available structures, the enzyme showing highest sequence similarity to SMFMO was a thioredoxin reductase from Thermus thermophiles, but similarity to PAMO and CHMORhodo was also observed.
BVMOs in natural catabolic processes
While in the past, the important role of BVMOs in the metabolism of compounds like acetone, bulky cyclic, bicyclic and aliphatic ketones, linear ketones and steroids was shown, recently the involvement of these enzymes in catabolic pathways was reported.40 One of the few examples of BVMOs that has been assigned a specific biosynthetic role and a defined substrate are the 1-deoxy-11-oxopentalenic acid-monooxygenases.41,42 These enzymes are involved in pentalenolactone D and neopentalenolactone D biosynthesis by three different Streptomyces species (Scheme 2). The gene clusters responsible for the whole metabolic pathway were cloned and sequenced. Pentalenolactone 7 is a sesquiterpenoid antibiotic, which is active against gram-positive and gram-negative bacteria as well as fungi because of its electrophilic epoxide moiety, which inactivates the glutaraldehyde-3-phosphate-dehydrogenase of those organisms. Pentalenolactone was isolated from numerous Streptomyces species. In 2009, a 13.4 kb gene cluster from Streptomyces avermitilis was cloned implicating 13 unidirectional ORFs. Among these genes was the putative flavin containing monooxygenase PtlE, which was recombinantly expressed in E. coli.41 Indeed, this enzyme turned out to be a FAD-dependent type I BVMO, catalyzing the Baeyer–Villiger oxidation of 1-deoxy-11-oxopentalenic acid 8 (Scheme 2). Surprisingly, the formation of the expected product pentalenolactone D 9 could not be observed. Instead, the formation of the regioisomer (and the more likely BVMO product) neopentalenolactone D 10 was found. That compound had never been isolated from Streptomyces or any other source before and it was concluded that the biosynthetic pathway of pentalenolactone debranches at the BVMO-reaction step and a new path was thereby identified (Scheme 2).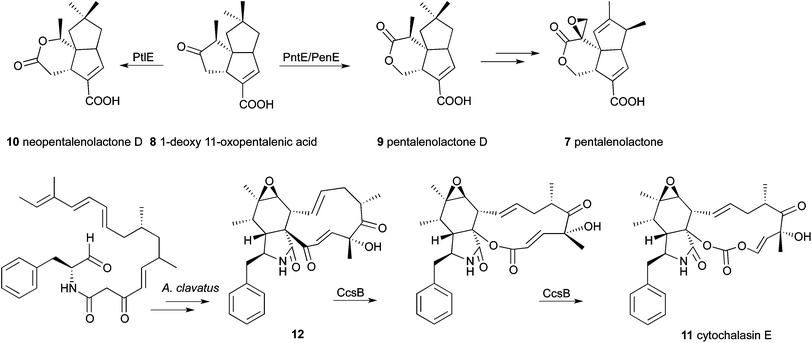 | ||
| Scheme 2 Involvement of BVMOs in the biosynthetic pathways of pentalenolactone and cytochalasin E. | ||
Since wild-type S. avermilitis showed the formation of new metabolites of sesquiterpenoids, but not pentalenolactone D itself, the gene clusters of two other representatives of Streptomyces species were investigated.42 The strains S. arenae and S. exfoliatus were known producers of the desired compound. The relevant ORFs of the pentalenolactone biosynthetic gene clusters of these strains were determined to be not only identical in organization, but also to exhibit a high degree of sequence identity. The PtlE-orthologous enzymes PntE and PenE showed about 80% similarity to the S. avermilitis protein PtlE. They were purchased as codon-optimized synthetic genes and overexpressed in E. coli to enable detailed investigations. For both enzymes, the exclusive FAD- and NADPH-dependent formation of the Baeyer–Villiger oxidation product pentalenolactone D (9) from 1-deoxy-11-oxopentalenic acid (8) was proven. PenE and PntE can therefore be considered as paralogues of PtlE, which catalyze the analogous oxidation of the same substrate, but yield the regioisomeric product. All three enzymes were found to be highly regiospecific. From mutational analyses, it was concluded that the N-terminal region, especially the region around the FAD-binding motif, influences the regiospecificity of the Baeyer–Villiger oxidation. Regarding the advantage of the availability of a catalyst for the formation of each regioisomer of a sesquiterpenoid, application of these enzymes in organic synthesis approaches seems promising.
Lately, researchers detected further strong hints for the contribution of another BVMO in a catabolic process. The intended study of the 30 kb ccs-gene cluster responsible for the biosynthesis of cytochalasin E (11) by Aspergillus clavatus NRRL 1 furthermore reports on BVMO activity in a eukaryote.43 Cytochalasins belong to secondary metabolites of the fungus and are of significant value because of their complex molecular structure and bioactivity (Scheme 2). The sequenced genome of A. clavatus NRRL 1 was searched for genes encoding a hybrid iterative type I polyketide synthase–nonribosomal peptide synthetase (PKS–NRPS). Next to a putative hit, which was identified, additional genes possibly involved in cytochalasin biosynthesis were observed. Based on the deduced gene functions of the ccs gene cluster, the biosynthetic pathway for cytochalasin E and K was proposed. It comprises, amongst others, six oxidative steps including two hydroxylations, one alcohol oxidation, one epoxidation and two Baeyer–Villiger oxidations. The enzyme responsible for the latter steps (CcsB) was assumed to be located directly downstream of the PKS–NRPS gene, because the ORF revealed about 25% identity to CHMOAcineto and CPMOComa. Moreover, it exhibits high sequence identity towards the recently characterized CPDMO from Pseudomonas sp. HI-70 (41%). It was found that CcsB contains the two intact conserved Rossmann fold motifs GxGxxG and GxGxxA, as well as the BVMO fingerprint. The 11-membered carbocyclic intermediate 12 resembles a very large BVMO substrate and the presumed ability of CcsB to convert it coincides with its close relation to CPDMO, which is capable of lactonizing C15 cycloketones. The fact that no additional genes encoding BVMO-like enzymes are located in the ccs cluster led the authors to the assumption that CcsB might be responsible for two consecutive Baeyer–Villiger oxidations resulting in compound 11. Since there are only slight hints available in the literature, experimental confirmation of this hypothesis is still required.
Novel synthetic applications
The overwhelming diversity of catalytic properties of BVMOs permits access to many different classes of valuable chemicals. An overview about the huge number of examples can be found in recent reviews.1–5 Recent studies reveal further powerful examples of the broad synthetic utility of these enzymes often leading to compounds one would not consider as products of a typical BVMO-mediated oxidation.The identification of CPDMO from Pseudomonas sp. HI-70 was discussed earlier in this perspective. As a valuable application, Fink et al. showed that CPDMO-catalyzed kinetic resolutions of racemic substituted cyclopentanones yielded full conversion to racemic lactones whereas it was possible to selectively oxidize only the (−)-enantiomer of 2-methylcyclohexanone to the normal lactone with E = 41.22 This behavior has only been observed for CDMO from Rhodococcus ruber CD4 (CDMORhodo) before. 2-Substituted cycloheptanones were not accepted by CPDMO. Regiodivergent transformation of the N-heterocyclic bicyclic ketone 13 led to formation of products distinct from a tested collection of ten BVMOs from various microbial origin and provided access to the antipodal Geissman–Waiss lactone (S,S)-14 as well as the abnormal product (R,S)-15 in a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture (Scheme 3). This means that the non-natural enantiomer of the naturally occurring alkaloids retronecine 16 and other necine bases are accessible via this chiral intermediate. Another non-conformity between CPDMO and CHMO-type BVMOs was observed for the conversion of menthone 17, where no regio-divergence was observed. Instead, both enantiomers (17a and 17b) were oxidized to the optical antipodes (18a and 18b). In conclusion, this approach discovered a number of novel biooxygenations extending the substrate scope within the BVMO family.
:50 mixture (Scheme 3). This means that the non-natural enantiomer of the naturally occurring alkaloids retronecine 16 and other necine bases are accessible via this chiral intermediate. Another non-conformity between CPDMO and CHMO-type BVMOs was observed for the conversion of menthone 17, where no regio-divergence was observed. Instead, both enantiomers (17a and 17b) were oxidized to the optical antipodes (18a and 18b). In conclusion, this approach discovered a number of novel biooxygenations extending the substrate scope within the BVMO family.
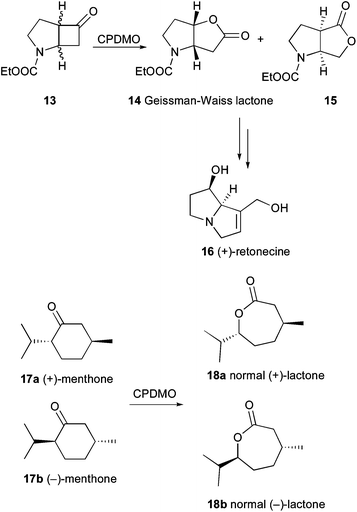 | ||
| Scheme 3 CPDMO from Pseudomonas sp. HI-70 provides access to the Geissman–Waiss lactone and is able to oxidize menthone. | ||
Aliphatic ketones
Until a few years ago, BVMOs were investigated mostly for the conversion of mono- and bicyclic ketones, camphor, a few arylaliphatic ketones and some steroids. More recently, it was discovered that BMVOs also catalyze the oxidation of aliphatic ketones to the corresponding esters. This also identified a possible physiological role of BVMOs. As was described for the BVMO from Pseudomonas fluorescens DSM 50106, a cascade of enzymes was found to be encoded in an operon including an alkane hydroxylase, an alcohol dehydrogenase, the BVMO and an esterase presumably being involved in the degradation of alkanes and thus enabling Ps. fluorescens growth on this carbon source.44 A similar pathway was later also found in Ps. putida KT2440.45It has been shown that not only simple aliphatic ketones are accepted as substrates, but that also β-hydroxy-substituted linear aliphatic ketones are oxidized in an enantioselective manner by eleven BVMOs of different bacterial origin and especially those of the CHMO-type.46 This observation is synthetically very useful as the kinetic resolution of these racemic compounds provides access to chiral β-hydroxyesters, which undergo acyl migration and ester hydrolysis by the whole-cell biocatalyst. This leads to the formation of optically pure 1,2-diols, which are valuable compounds in the synthesis of polyesters and antimicrobial agents. Moreover, the enantioconvergent conversion of racemic substrates by different enzyme candidates was observed.
Recently, the potential of BVMOs to form the abnormal ester of N-protected β-amino ketones was described. Coupling of a lipase for hydrolysis of the resulting ester provided access to enantiopure β-amino acids under mild reaction conditions46 (Scheme 4). This new enzymatic route also grants access to N-protected β-amino alcohols. In this recent approach, whole-cell experiments with 16 BVMOs from various bacterial strains were investigated for their acceptance of protected 5-amino-3-one as substrates in the kinetic resolution mode. This revealed that four enzymes (a CHMO from Arthrobacter BP2 (CHMOArthro), a CHMO from Brachymonas petroleovorans (CHMOBrachy) and CHMOXantho as well as CDMORhodo) showed activity. Interestingly, the non-protected amino alcohols were not converted by any enzyme. When biocatalysis was performed in 24-well microtiter plates, all four enzymes formed roughly 1:1 ratios of normal and abnormal products and all were obtained with excellent enantioselectivity with E-values > 200 except for the formation of the normal ester by CDMO, where 81% ee was measured. Interestingly, the ratio of regioisomers formed turned out to be dependent on reaction conditions, which amongst other factors was explained by the availability of oxygen as higher conversion of the substrate was observed, when the reaction was performed under conditions with improved oxygen supply. Due to their pharmaceutical relevance in the synthesis of β-peptides, alkaloids, terpenoids and β-lactam antibiotics, β-amino acids represent desirable compounds for organic synthesis. Because of their enhanced stability towards human proteolytic enzymes, these compounds are particularly interesting for the design of drugs.
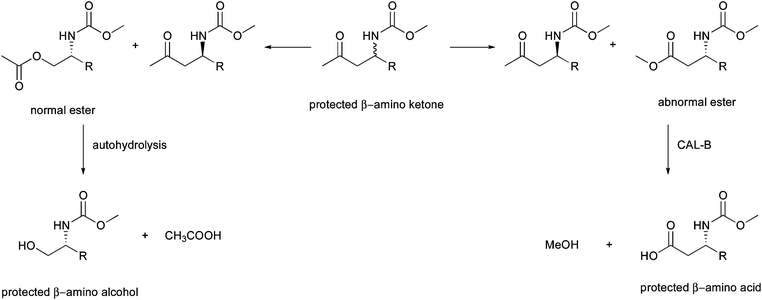 | ||
| Scheme 4 Enzymatic Baeyer–Villiger oxidation of protected β-amino ketones provides access to β-amino alcohols and β-amino acids. | ||
This collection of enzymes was also used in a subsequent study to investigate the formation of β-amino alcohols, which are of great pharmaceutical interest because this motif occurs in many different drugs. β-Amino alcohols are difficult to access in enantiopure form by chemical means and only a few enzymatic methods for the synthesis of these compounds have been described before. Rehdorf et al. investigated the conversion of linear aliphatic, branched linear and arylaliphatic β-amino ketones.47 Whole cell preparations of ten BVMOs converted these racemic N-protected compounds. Throughout the linear aliphatic substrates, the CHMO-type enzymes preferred the medium chain length (C8) and conversion decreased dramatically when the chain length was increased to 12 carbon atoms. A complementary trend was observed for HAPMOACB and CDMO, which have been known to prefer structurally demanding ketones. Detailed analyses of the relationship between time and enantiomeric excess at approximately 50% conversion revealed that the C8 aliphatic β-amino ketone was converted the fastest by CHMOBrachy with E > 200. Similar results were obtained for CHMOXantho and CDMORhodo for chain lengths of 10 carbons. The branched chain aliphatic β-aminoketones were converted with moderate activity, but enantioselectivity was poor except for CHMOArthro (E > 200). For these substrates it was observed that the opposite enantiomer is converted by almost all tested enzymes when the side chain and the keto-function were separated by one more carbon. For the aryl-aliphatic substrate, high activity was observed for almost all enzymes, but only PAMO and cyclohexanone monooxygenase from Brevibacterium sp. HCU (CHMOBrevi) showed good enantioselectivity. An alignment of the amino acid sequences of seven enzymes active towards arylaliphatic ketones led to the identification of a loop segment, which occurs in PAMO, CDMO and CHMOBrevi and is missing in the other CHMOs. The two amino acids reduce the size of the binding pocket in PAMO and this could therefore explain the high enantioselectivity observed.
The β-amino alkylesters formed by the BV-oxidation underwent spontaneous hydrolysis due to the increasing pH in the whole-cell system and hence the N-protected optically active β-amino alcohol became accessible. Regarding the possibility to regulate which product enantiomer will be formed by choice of the appropriate enzyme as catalyst, BVMOs were shown in the recent work by Rehdorf et al. to be an essential tool in the synthesis of chiral compounds and even offer access to unexpected compounds like 1,2-diols, β-amino alcohols or β-amino acids.
The strategy of subjecting the entire BVMO collection to a set of compounds was used by the group of Mihovilovic who thus succeeded in the identification of two enzymes for the kinetic resolution of 2-substituted cycloketones.48 The recovered substituted chiral δ-valerolactones and ε-caprolactones are known as flavor and fragrance compounds. They have been identified in plants like jasmine ((R)-23), agaves ((S)-23) as well as natural mango aroma (Scheme 5). In the screening step seven enzymes from known cycloketone-converting BVMO families were identified, which readily transformed substrates 19–22 into the expected lactones with the same regio- and enantiopreference at 50% conversion. CHMOArthro showed excellent enantioselectivity in the resolution of 19–21 and CDMORhodo for 22. This work resembles the first example of BVMOs employed in the preparation of aroma lactones.
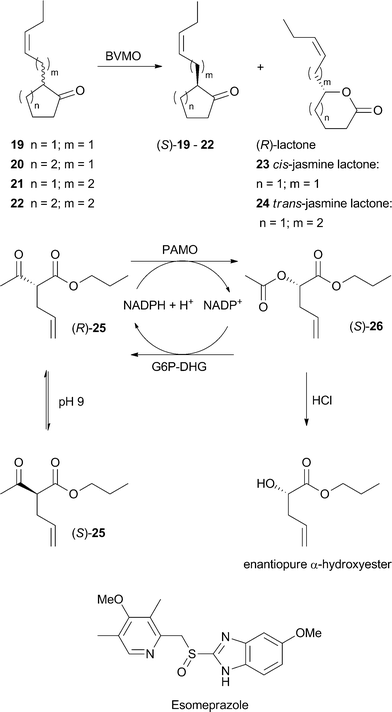 | ||
| Scheme 5 Application of BVMOs in the synthesis of aroma compounds, DKR of α-substituted β-keto esters and production of the drug Esomeprazole. | ||
The pallet of compounds contrivable by BVMOs was recently widened by a dynamic kinetic resolution (DKR) approach. In this study, aliphatic acyclic α-substituted β-keto esters were subjected to PAMO, its mutant M446G and CHMOAcineto49 with spontaneous racemization of the starting material at pH 9. Although the BMVOs chosen normally exhibit substrate preferences for aromatic ketones, the aliphatic acyclic racemic α-alkyl β-ketoesters were also accepted albeit with low conversion by the PAMO mutant. In a DKR with 25 complete conversion to 26 was found after 24 h (Scheme 5). Selective hydrolysis of the diesters was performed chemically using catalytic amounts of hydrochloric acid and resulted in enantiopure α-hydroxyesters, which are widely applicable in the pharmaceutical production of anticancer drugs and antibiotics as well as in the food industry.
Heteroatom-substituted compounds
In addition to the broad number of compounds accessible with BVMOs through the oxygenation at a keto moiety, the oxygenation of heteroatoms like sulfur, nitrogen, phosphorus, boron and selenium widens the applicability of this enzyme class. In one approach, achiral aromatic and vinylic boron compounds as well as racemic ones have been evaluated as target substrates where oxidation aiming at the carbon–boron bond would afford the corresponding alcohols by elimination of boronic acid.50 Five different acetophenone derivatives bearing boron substituents at the m- or p-position were employed. PAMO, its mutant M446G, HAPMOACB and CHMOAcineto were chosen as biocatalysts. PAMO and its mutant were equally chemoselective for the boron oxidation of all substrates affording the corresponding phenols, but mutant M446G showed lower activity. In HAPMO-catalyzed reactions boron oxidation as well as BV-oxidation was observed. CHMO showed high chemoselectivity in favor of boron oxidation, but low activity. Using this enzyme, only the 3- or 4-hydroxyacetophenones were afforded, but at poor conversions. Moreover, the oxidation of vinyl boron compounds was explored in that work to evaluate the chemoselectivity between boron oxidation and a possible epoxidation reaction, which has previously been described.51,52 Thereby, aliphatic vinylic boron compounds turned out to be no substrates for the chosen catalysts, but aromatic substrates of this class were oxidized exclusively at the boron and no epoxidation was observed at all. M446G was furthermore applied to evaluate the enzymatic kinetic resolution of a chiral boron-compound. It was observed that exclusively the (S)-borane was transformed into the corresponding (S)-alcohol with high enantiomeric excess. These results are valuable since boron-containing compounds are versatile intermediates in synthetic organic chemistry. The same is true for organo selenoxides, which find application as mild oxidation reagents and catalysts in hydrogen peroxide activation, therefore a further study was aimed at evaluating the chemoselectivity of PAMO in the biooxidation of organoselenium acetophenones.53 Conversion of acetophenone derivatives bearing selenide substituents at all three possible positions on the aromatic ring yielded the corresponding selenoxides in high conversion after 24 h while PAMO was chemoselective by only catalyzing selenium oxidation.In addition, a Baeyer–Villiger monooxygenase was engineered by the company Codexis Inc. for a sulfoxidation to yield the drug Esomeprazole (Scheme 5). Protein engineering was used to invert the enantiopreference and to improve the enzyme with respect to activity, stability, and chemoselectivity.54
Limitations of BVMO-catalyzed reactions
Although a variety of new biocatalysts have been identified during recent years and novel synthetic applications have been shown, still a number of drawbacks have to be overcome to enable the use of most BVMOs on an industrial scale. One of the major obstacles is the limited stability, low substrate and hence product concentrations, adequate oxygen transfer as well as tolerance of organic cosolvents.55 To circumvent these limitations several strategies have been developed during the last years.Optimization of biotransformation conditions
The addition of a water miscible organic solvent to improve substrate solubility is often encountered with reduced enzyme stability. In a recent study the stability and activity of PAMO and CHMOAcineto in the presence of organic solvents such as methanol, ethanol, 1,4-dioxane, acetonitrile and 1,1,1-trifluoroethanol were analyzed.56 PAMO turned out to be significantly more stable than CHMOAcineto concerning the percentage of solvent added as well as the long-term stability at given concentrations. Interestingly, the addition of 20% methanol resulted in an about five-fold increase of PAMO activity while CHMOAcineto activity was only 1.2-fold higher at a maximum of 2% methanol. Fluorescence data and circular dichroism analyses indicated that the decrease in catalytic activity for both enzymes at increasing concentrations of organic solvent was caused by a loss in tertiary and secondary structures. Computational comparison of PAMO and CHMO structures identified the number of salt bridges in both enzymes, which are known to increase the protein thermal stability. So the higher amount of salt bridges in PAMO (41) compared to CHMOAcineto (31 or 20 depending on whether the closed or the open form model was used) also seems to enhance the stability of the BVMOs in water–organic solvent mixtures.56 Another approach aimed at analyzing substrate acceptance and enantioselectivity of the PAMO mutant M446G in the presence of varying concentrations of hydrophilic organic solvents.57 In the oxidation of benzyl methyl sulfide, the addition of 10% PEG or MeOH led to an almost complete formation of the sulfoxide whereas in EtOH, iPrOH or CH3CN the sulfone was the major product. Cyclohexyl propyl sulfide and rac-2-phenyl-3-heptanone could only be oxidized to the corresponding sulfone and ester in the presence of an organic solvent. In a DKR, the addition of 5% MeOH enabled up to 90% conversion of rac-3-phenylbutan-2-one with Lewatit MP62 (89% ee of the product) and for various benzylketones also high yield and optical purity could be achieved.57The addition of water-immiscible organic solvents creates a biphasic system, which on the one hand acts as a substrate reservoir and on the other hand as an extraction medium for in situ product removal from the aqueous phase. Thus, both the substrate and the product concentration can be kept below inhibitory levels and therefore the biocatalyst can be stabilized significantly by the addition of the cosolvent for a longer period of time.58
Alternatively, ionic liquids (ILs) can be used instead of organic solvents. The advantage of ILs is that the polarity, hydrophobicity, viscosity and solvent miscibility can be tuned by altering the type of cation and anion. This allows the design of media for different purposes.59 It was found that besides their expected solvent properties, ILs can have a particular impact on enzyme activity and selectivity. In a recent study the PAMO-catalyzed kinetic resolution of rac-α-acetylphenylacetonitrile was investigated. Employment of the IL [bmp]PF6 reduced the formation of the by-product phenylacetonitrile from 56 to 3% while the yield of the BVMO product could be increased from 4 to 48% with excellent optical purity of >99% ee. Additionally, the space-time-yield could be improved by increasing the substrate concentration from 10 to 120 mM. Unfortunately, PAMO was inactivated in the presence of IL after 72 h.59
In addition, it was shown for PAMO that also the buffer system and the ionic strength had a strong influence as exemplified in the kinetic resolution of rac-3-phenylbutan-2-one. Tris- and phosphate buffers gave best results leading to fast conversion of the substrate and an excellent E = 120. Other buffer systems either led to faster product formation, but reduced enantioselectivity or extremely slow conversion.60 This phenomenon might be explained by neutralization of electrostatic interactions on the protein surface due to high salt concentrations that finally affect the protein structure.61
As most BVMOs require reduction equivalents and the stoichiometric addition of the cofactor NAD(P)H is expensive, an efficient cofactor regeneration system is needed. Besides the use of a whole cell system with ‘integrated’ cofactor recycling by the addition of glucose, the PAMO-catalyzed oxidation of phenylacetone was explored with isolated enzymes coupled to several enzymatic cofactor recycling systems such as glucose/GDH,62 glucose-6-phosphate/G6PDH,63 iPrOH/TBADH,64 sodium phosphite/PTDH or using a fusion protein (CRE2-PAMO).65–68 The use of glucose dehydrogenase (GDH) at pH 8.0 and 30 °C and glucose-6-phosphate dehydrogenase (G6PDH) at pH 9.0 and 30 °C exhibited highest productivities (∼40 mmol mL−1 h−1) similar to the phosphite dehydrogenase (PTDH) system. The alcohol dehydrogenase (TBADH) gave poor results. Highest total turnover number and turnover frequency were observed in the presence of only 2 μM NADPH. Interestingly, the PTDH and the G6PDH systems also gave higher selectivity (E > 100 for rac-3-methyl-4-phenylbutan-2-one) but rather slow conversion, whereas with GDH faster conversion but lower selectivity was observed.60 Similar results were observed for the oxidation of thioanisole to the corresponding (S)-methyl phenyl sulfoxide.
Substrate feeding and product removal approaches
The use of whole cells of the microorganism expressing the BVMO of interest has the advantage that cofactor regeneration is substantially facilitated. However, whole cell biocatalysts are more sensitive to the addition of cosolvents and especially high substrate or product concentrations, which also affect the performance of isolated enzymes. For example, CHMOAcineto has been shown to be inhibited by concentrations of 3 mM of rac-bicyclo[3.2.0]hept-2-en-6-one as the substrate and 36 mM of the resulting two lactones, respectively.69 A couple of further examples were published by the group of Woodley70 and are covered in a recent review.1One strategy to circumvent biocatalyst inactivation by critical substrate concentration is the continuous feeding aiming to maintain the substrate concentration below an inhibitory level.71,72 A further strategy focuses on an appropriate in situ removal of the product formed.73 Combining both approaches leads to the in situ SFPR (substrate feed and product removal) concept that has been utilized in several studies.74,75 While most published examples employ the CHMOAcineto74,76 only one example used HAPMO from Ps. putida JD1.77 In this study the scale-up as well as the in situ SFPR strategy were investigated for the kinetic resolution of 3-phenyl-2-butanone, which served as a chiral model substrate for this enzyme.78 First attempts with 1.4 mM substrate gave 45.6% conversion with excellent optical purity of the product (99.2% ee) and E > 100.77 Already at 5.4 mM the conversion dropped drastically due to the lack of proper oxygen supply, which could be simply overcome by changing the reaction vessel. In order to further increase the substrate concentration, various adsorption resins were investigated and Dowex® Optipore® L-493 and Lewatit® VP OC 1064 MD PH gave the best results if an optimal ratio between the resin and the substrate is ensured. This resulted in 39% (Dowex®) and 45% (Lewatit®) conversion at substrate concentrations >26 mM. Hence, variation of type and concentration of the resin enabled optimal conditions avoiding inhibition at higher substrate and product levels.
Immobilization of BVMOs
As outlined above, the application of BVMOs in industrial processes is still hampered by several factors. Immobilization of the biocatalysts (Table 3) can circumvent stability issues and facilitate enzyme recovery, but can also enable easier cofactor regeneration if the recycling enzyme is co-immobilized. Although free enzymes can be recycled by ultrafiltration, in the case of BVMOs the low mechanical stability usually prevents this method as shown by Zambianchi et al. for the oxidation of bicyclo[3.2.0]hept-2-en-6-one (5 g L−1).64 125 U of CHMO and 125 U of the alcohol dehydrogenase from Thermoanaerobium brockii (TBADH) were used in repeated batches (each 24 h) with recycling by membrane filtration. After three cycles only 40% conversion was reached and it was shown that this activity loss was due to the inactivation of CHMOAcineto during the process. As is typical in enzyme immobilization, the identification of the best carrier and immobilization method is a rather tedious trial and error task. The first reported immobilization of a BVMO was the entrapment of the CHMOAcineto in a polyacrylamide gel.79 The cofactor regeneration was realized with the G6PDH system, which was separately entrapped in the polyacrylamide gel. The immobilized preparations were used for the biooxidation of different cyclic ketones with concentrations ranging between 40 and 100 mM in a 1 L reaction volume. Within five to ten days it was possible to isolate between 75 and 89% of pure lactones. For the conversion of 2-norbornanone the retained activity of CHMO was 77% and for G6PDH 80%. Co-immobilization of CHMO and G6PDH was tried by attachment on glyoxyl-agarose coated with polyethyleneimine (PEI).80 The immobilized CHMOAcineto showed a broader pH profile in the conversion of 2-oxocyclohexyl acetic acid to the corresponding lactone and the temperature optimum was increased by 5 °C, however the method was not very efficient as only 0.26 U gSupport−1 could be attached to the surface and large amounts of NADPH were required. The activity of the immobilized cofactor regenerating enzyme was not experimentally confirmed. Another co-immobilization of CHMOAcineto was done with the TBADH on Eupergit® C. The immobilized enzymes showed good stability during oxidation of thioanisole (80% conversion after 17 batches, each 24 h) or bicyclo[3.2.0]hept-2-en-6-one (80% conversion after 4 batches, each 24 h). Recently, PAMO was immobilized with G6PDH on a polyphosphazene support and used for the oxidation of phenylacetone,81 but the recovered activity on the support and the stability were rather low. Another example for PAMO used encapsulation in peroxisomes.82 The authors could show that the fusion enzymes CRE2-PAMO showed higher activity than the single co-encapsulated enzymes. Nevertheless, the encapsulated CRE2-PAMO showed decreased activity compared to the soluble enzymes, which was explained by diffusion problems. Until now no immobilization system could be identified, which leads to a highly active and stable biocatalyst with satisfying performance. Whole cell immobilization was shown for E. coli cells expressing CPMOComa in polyelectrolyte complex capsules (PEC) used for the oxidation of 8-oxabicyclo[3.2.1]oct-6-en-3-one.83 The encapsulation process was visualized using confocal laser scanning microscopy (CLSM) and around 94% of cells were viable. The encapsulated cells showed significant improvement of storage stability, but a 5 times lower activity (0.12 U g−1 cells) compared to free cells. During biooxidation, the immobilized cells showed the same conversion (over 90%) of the ketone after 48 h with comparable enantioselectivity to the free cells, but reusability was not reported. For the encapsulation of E. coli cells with CHMOAcineto in PEC a recycling and storage stability study showed that the cells showed high stabilization benefits due to the encapsulation. The cells could be reused for 14 repeated biotransformations of rac-bicyclo[3.2.0]hept-2-en-6-one (each 12 h) with a starting conversion of 77% in the first and 75% conversion in the 14th cycle. The storage ability of the cells was increased drastically with conversions of 80% after 60 days and 50% after 91 days. These approaches show the potential for encapsulation of BVMO expressing whole cells ensuring a high stabilizing effect. However, until now encapsulation in PEC matrixes is limited by the low activity of the entrapped cells, which appears to be unsuitable for industrial application. Diffusion problems and limited oxygen supply could be one explanation for these low activities.| Enzyme | Amount of biocatalyst | Substrate | Conc. (mM) | Support (binding mode) | Reaction time | Conv. (%) | Comment | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Whole cells. b Oxygen aeration. c Bubble free oxygen aeration and continuous flow reactor. d Reaction volume ≤2 ml; cofactor recycling with e G6PDH (glucose-6-phosphate dehydrogenase) or f alcohol dehydrogenase from Thermoanaerobium brockii; n.r. not reported. | ||||||||
| CHMOAcinetoa | 50 U CHMO | 2-Norbornanone | 100 | Polyacrylamide | 5 d | 100 | 30% immobilization yield (by protein concentration); CHMO recovered with 77% activity (after complete conversion of 2-norbornanone); G6PDH entrapped separately | 79 |
| L-Fenchone | 100 | Gel (entrapping)e | 8 d | 100 | ||||
| 100 U G6PDH | D-Fenchone | 100 | 10 d | 100 | ||||
| (+)-Camphor | 50 | 10 d | n.r. | |||||
| (+)-Dihydrocarvone | 40 | 10 d | n.r. | |||||
| CHMOAcineto | 10 U | Thioanisole | 38d | Eupergit® C | 24 h | 100 | 80% conversion in 17th cycle (thioanisole); 80% in 4th cycle (bicyclo[3.2.0]hept-2-en-6-one); half-life at 25 °C increased 2.5-fold | 64 |
| Bicyclo[3.2.0]hept-2-en-6-one | 46d | (Covalent)f | 24 h | 100 | ||||
| CHMOAcineto | n.r. | (2-Oxocyclohexyl) acetic acid | n.r. | PEI coated glyoxyl-agarose (adsorption)e | 24 h | 67 | G6PDH activity experimentally not confirmed; Topt +5 °C; pHopt broader; γ-irradiation improves stability; 0.26 U g−1 for cyclohexanone | 80 |
| PAMO | 20 mg | Phenylacetone | ∼9.5d | Polyphosphazene (covalent)e | 24 h | n.r. | Low recovered activity on support; 80% activity loss after 5 cycles; co-immobilization: 3.2 U g−1 | 81 |
| CRE2-PAMO | n.r. | Phenylacetone | 2.5 | Peroxisome (encapsulation)e | 15 h | 100 | CRE2-PAMO higher activity then co-encapsulation of both enzymes; activity reduced | 82 |
| CPMOComaa | n.r. | 8-Oxabicyclo[3.2.1] oct-6-en-3-oneb | 5.7 | Polyelectrolyte complex capsule (encapsulation) | 48 h | 91 | 5-fold lower activity compared to free cells; 94% cells viable after encapsulation; 0.12 U g−1 cells; storage stability improved | 83 |
| CHMOAcinetoa | rac-Bicyclo[3.2.0]hept-2-en-6-onec | 1.85 | Polyelectrolyte complex capsule (encapsulation) | 12 h | 77 | 0.12 U g−1 cells; 14th cycle; storage stability improved | 108 | |
Recently, a new expression system in Corynebacterium glutamicum for CHMOAcineto was established, overcoming substrate inhibition of cells and enabling high productivity during fed batch biotransformation.84 The high conversion was explained by a more efficient cofactor regeneration system.85 To circumvent diffusion problems through the cell wall, permeabilization was achieved with ethambutol.86 For molecules >170 g mol−1, the affinity could be increased by 30%, which indicates a permeabilized cell wall. These new host cells hence appear to be a more suitable system to overcome the low activity of entrapped cells.
Crystal structures of BVMOs
Since 2011, different 3D-structures of PAMO88 and the newly characterized OTEMO were solved.11,12 Orru and coworkers crystallized PAMO during different steps of BVMO catalysis with a focus on the structural mechanism of the oxidation process (Table 4, Fig. 1). The snapshots provided deep insights into the PAMO structure with bound FAD/NADPH and the enzyme in its oxidized and reduced form. For the oxidized wild-type with bound FAD and NADP+, the authors predicted that NADPH binds near the flavin N5 atom for hydride donation as it was described for CHMOAcineto.89 Then the NADP+ slides over to the flavin and stabilizes the flavin (hydro)peroxide. With a reduced form of the wild-type enzyme they gained insight into the flavin-peroxide formation. The carboxyamide group from NADP+ forms a H-bond to the N5 atom of the reduced flavin to prevent intermediate collapse of the flavin (hydro)peroxide. In contrast, in the oxidized form the crucial R337 residue forms H-bonds to the nicotinamide and interacts with D66. In the reduced enzyme state R337 moves to the pyrimidine moiety of the flavin ring and can interact with the negatively charged reduced flavin. Due to this movement Orru and coworkers predicted that flavin is accessible to O2 to form the flavin (hydro)peroxide. The flavin (hydro)peroxide shifts back and interacts with the nicotinamide – because of the loss of the negative charge – and the active site becomes accessible. With the mutant D66A the authors could show that R337 directs the substrate into the active site. In the snapshot (pdb-code: 2YLT) they demonstrated that R337 had two functions, which is first the increased nucleophilic attack against the flavin peroxide and second that it compensates the negatively charged Criegee intermediate. Mutant R337K confirmed that the enzyme in its oxidized form can still form a stable Criegee intermediate. In its reduced form, the mutant could still bind 2-(N-morpholino)-ethanesulfonic acid (MES) despite the lack of the guanidine group. Mutant M446 showed a widened pocket, which explains the broader substrate specificity and conversion of aromatic compounds.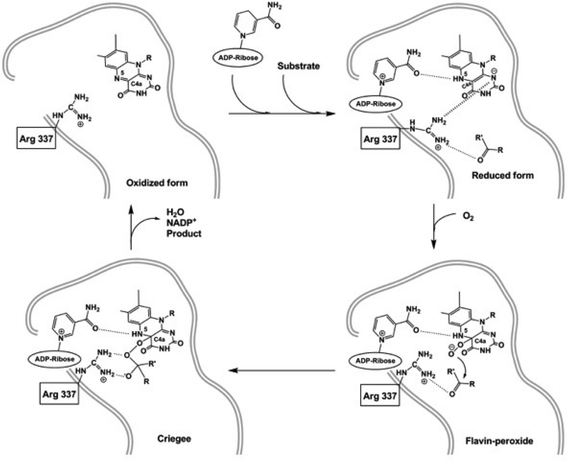 | ||
| Fig. 1 Mechanism of PAMO-catalyzed Baeyer–Villiger oxidation as derived from 3D structure analysis. | ||
| Enzyme | Pdb-code | Resolution (Å) | Comment | Ref. |
|---|---|---|---|---|
| a FAD+. b NADP+. c 2-(N-Morpholino)-ethanesulfonic acid. d Oxygen. e 1,2-Ethanediol. | ||||
| PAMO | 1W4X | 1.7 | 2 domains (one for FAD and one for NADP+ binding; active site in cleft of domain interface); R337 re side to flavin, R337 in “IN” and “OUT” conformation | 17 |
| 2YLR , | 2.26 | Oxidized form; structure shows NADP+ binding and its stabilization of flavin-(hydro)peroxide; R337 interacts with NADP+ and side chain D66 | 88 | |
| 2YLS , | 2.26 | Reduced form; structure shows flavin-peroxide formation; carboxyamide group of NADP+ makes H-bond with N5 from reduced FAD to prevent reaction with flavin-peroxide; R337 interacts with negatively charged reduced flavin favoring accessibility for O2 | 88 | |
| 2YLT , , | 2.65 | MES in the active site is shown to be in direct contact with R337 and the ribose group of NADP+ | 88 | |
| PAMO N337K | 2YLW , , | 2.9 | Mutant can still bind MESc, but cannot interact with NADP+, D66 and ligand simultaneously | 88 |
| 2YM1 , , | 2.28 | Oxidized form; K337 interacts with carboxamide group of NADP+ and side chain of D66 | 88 | |
| 2YM2 , | 2.70 | Reduced form; K337 moves to the flavin to a similar conformation as R337 in WT | 88 | |
| PAMO D66A | 2YLX , , | 2.20 | Mutant showed lower kcat for NADPH; negative charge facilitates positioning of NADPH | 88 |
| PAMO M446G | 2YLZ | 2.00 | Mutant accepts aromatic compounds; showed no conformational changes, but widened pocket | 88 |
| MtmOIV | 3FMW , | 2.89 | Dimer; R52 (similar to R337 in PAMO), but in si side orientation to flavin; class A flavoprotein monooxygenase; needs peroxyflavin intermediate | 90 |
| CHMO closed | 3GWD , | 2.30 | 2 domains (one for FAD and one for NADP+ binding); R329 (similar to R337 in PAMO) pushes nicotinamide head deeper to stabilize peroxyflavin and “Criegee” intermediate (causing “sliding” of NADP+); represents enzyme in post-flavin reduction state; structure confirms novel role of BVMO sequence motif as it coordinates domain movements during catalysis | 89 |
| CHMO open | 3GWF , | 2.20 | R329 in “OUT” conformation (similar to R337 in PAMO); structure shows final step of NADP+ release in the catalytic cycle | 89 |
| 3,6-DKCMO | 2WGK | 2.00 | Structure determined only by non-crystallographic symmetry (NCS) exhaustive search | 91 |
| OTEMO | 3UOV | 2.05 | Dimer | 11 |
| 3UOX | 1.96 | 11 | ||
| 3UOY , | 2.00 | 11 | ||
| 3UOZ , | 2.41 | 11 | ||
| 3UP4 , | 2.80 | Closed form | 11 | |
| 3UP5 , | 2.45 | 11 | ||
Assay systems to identify BVMOs
In order to allow fast and reliable identification of novel BVMOs or variants within protein engineering derived mutant libraries, it is crucial to have high-throughput assays available. Assay systems to measure product formation from BVMO-catalyzed reactions are based either on a pH shift after hydrolysis of the resulting ester or lactone, respectively, or on the formation of chromo- or fluorogenic compounds liberated after cleavage of the resulting ester BVO-product. In 2002 Littlechild et al. introduced an assay employing pig-liver esterase (PLE) to induce a pH shift that occurs in a non- or weakly-buffered system through a pH-indicator.92 However, this method was only applicable to washed cell suspensions, as various factors in a whole cell system can lead to a change in pH, which again can entail erroneous results. A fluorogenic assay was based on the detection of umbelliferone (7-hydroxycoumarin) formed from 4-oxopentyl umbelliferyl ether by a BVMO reaction and subsequent oxidation of the formed alcohol, which was first reported in 2003.93 Umbelliferone also served as a reporter in another assay in which the oxidation product of 2-coumaryloxy ketones was subsequently cleaved by PLE to release the fluorescent dye.94 Both assays require the multi-step synthesis of the non-commercially available starting material and acceptance of the bulky substrates by the BVMO. The successful adaptation of an assay based on adrenalin conversion, initially described by Wahler and Reymond,95 was used by our group to identify mutants of a BVMO from Pseudomonas fluorescens DSM 50106 that showed enhanced conversion and enantioselectivity in the kinetic resolution of 4-hydroxy-2-decanone.96 After BVO and subsequent hydrolysis of the formed ester by an esterase, a 1,2-diol is formed, which can react with NaIO4. This assay operates through back titration of non-reacted NaIO4 with adrenaline yielding the chromophore adrenochrome. The method works in microtiter plates (MTPs), but is unsuitable for the determination of enzyme kinetics as it allows only endpoint measurements. More recently, an assay based on monitoring cycloalkanone consumption was shown to be applicable for qualitative screening as well as quantitative activity determination.97 In alkaline solution, the enolizable ketone forms a colored complex with 3,5-dinitrobenzoic acid, which can be used to follow the decrease in absorption in case the ketone is oxidized. The method was shown to work for cycloalkanones with ring sizes between C4 and C7, but absorbance of the color decreased with the size of the cycloketone ring. Very recently, we described an assay based on the BVO of p-nitroacetophenone.98 The resulting acetate is subsequently hydrolyzed by an esterase or NaOH to yield p-nitrophenolate that can easily be quantified spectrophotometrically at 410 nm. The assay principle was applied to whole E. coli cells containing recombinant BVMO, crude cell extract as well as to purified enzyme. Screening of mutant libraries of the 4-hydroxyacetophenone monooxygenase (HAPMO) from Pseudomonas putida JD1 using this assay could identify more active enzyme variants.Protein engineering to tailor-design BVMOs
Enzymes in nature hardly meet the demands imposed by an industrial application. For this, biocatalysts can be adapted to their required characteristics (such as substrate scope and concentration, selectivity, temperature, pH, stability) by methods of protein engineering.99–101 In case the three-dimensional structure of an enzyme is available, rational protein design is often the method of choice, although it is still challenging to predict the effects of a distinct mutation. Alternatively, methods of directed evolution are used in order to improve the protein by random mutagenesis or libraries created by simultaneous saturation mutagenesis, which need to be screened with an appropriate assay to identify desired variants. A summary of the most recent protein engineering examples for BVMOs is given in Table 5. These demonstrate that activity, enantioselectivity, substrate range and stability of BVMOs could be successfully improved creating more versatile enzyme variants.| Target enzyme | Method | Mutations | Desired objective | Results/comments | Ref. |
|---|---|---|---|---|---|
| CASTing: Combinatorial Active Site Saturation Testing; SLIC: Sequence and Ligation Independent Cloning. | |||||
| PAMO | Saturation mutagenesis using degenerate primers | S441A/A442W/L443Y/S444T | Increasing the activity toward a substrate that is hardly converted by WT PAMO | Alignment of WT PAMO with seven other BVMOs, limited number of amino acids at positions 411–444, mutant screening on rac-2-phenyl-cyclohexanone identified quadruple mutant with E = 70 favoring the (R)-enantiomer in contrast to (S)-selective wild-type. This mutant also oxidized 2-(4-chlorophenyl)-cyclohexanone with excellent selectivity (E > 200). | 102 |
| PAMO | CASTing, saturation mutagenesis | “Second sphere” residue P440N | Expanding the substrate scope; higher E-values, maintenance of thermostability | Screening with rac-2-ethylcyclohexanone (not converted by wild-type enzyme) resulted in five highly active hits, which were analyzed in kinetic resolutions using various 2-substituted cyclohexanones; best mutants converted all cyclic ketones with E > 200; some mutants gave formation of ‘abnormal lactone’ with rac-bicyclo [3.2.0]hept-2-en-6-one. | 103 |
| PAMO | Site directed mutagenesis, saturation mutagenesis using NDT codon degeneracy | Q93N/P94D | Expanding the substrate scope | Introduction of distal mutations at positions Q93/P94 induced allosteric interactions between the N-terminal region of an α-helix (Ala91–Glu95) and the loop segment Tyr56–Tyr60 (FAD-binding domain) causing movement of the loop segment Trp177–Glu180 (NADP-binding domain). A double mutant Q93N/P94D gave good to excellent selectivity in the conversion of 2-substituted cyclohexanones and desymmetrization of 4-substituted cyclohexanones. MD simulations suggested new H-bonds (Asp94/Arg59 and Trp57/Trp177) and a strong salt bridge between Asp94 and Arg59. | 104 |
| PAMO | Site directed mutagenesis | H220N, H220Q, K336N | Changing the cofactor specificity to NADH | 3-fold increase of the catalytic efficiency of mutants using NADH as reduction equivalent compared to wild-type enzyme, mutant K336N showed a significantly increased E-value in the kinetic resolution of rac-3-methyl-4-phenylbutane-2-one for both NADH and NADPH. | 105 |
| PAMO | CASTing, site directed mutagenesis | V54, I67, Q152, A435 | Expanding the substrate scope | Comprehensive inspection of the active site of PAMO (crystal structure) and CPMO (homology model based on PAMO). Exchange of various active site amino acid residues in PAMO to its counterparts in CPMO. Single and multiple mutants (15 each) were analyzed in oxidation reactions of 14 different ketones and sulfides. Amino acids V54, I67, Q152, and A435 in PAMO contributed to the substrate specificity and enantioselectivity; a partially inverted enantioselectivity similar to CPMO/CHMO was observed too. | 106 |
| PAMO | Structure-inspired subdomain exchanges by the SLIC method | Chimeric BVMOs | Expanding the substrate scope | Blending of the substrate specificities of sequence-related BVMOs (STMO, CHMO and a putative BVMO from a metagenome screening effort107) into PAMO. Construction of three chimeras (PASTMO, PACHMO and PAMEMO1) consisting of 106 C-terminal amino acid residues of PAMO exchanged by homologous regions of the other enzymes. Characterization of all chimeras (melting temperature, substrate acceptance (using thioanisole, benzyl phenyl sulfide, rac-bicyclo-[3.2.0]hept-2-en-6-one, rac-2-phenylcyclohexanone and progesterone) and selectivity). All chimeras exhibited novel catalytic activity, especially concerning regio- and stereoselectivity, but not all activities from the parent BVMOs could be introduced into the constructs. Thermostability was significantly increased for all chimeras compared to parental BVMO. | 107 |
| CHMOAcineto | Site directed mutagenesis, saturation mutagenesis | M5I, M291I, C330S, C376L, M400I, M412L, M481A, C520V | Design of mutants with enhanced oxidative stability and thermostability | Replacement of Met and Cys residues by amino acids with small hydrophobic side chains (Ile, Leu, Ala) present in PAMO and CHMORhodo. Mutation C376L afforded the highest improvement in oxidative stability, while an M400I mutation resulted in the largest increase in thermal stability. Recombination of all improved mutants yielded two mutants with significantly increased oxidative and thermostability. While the wild-type CHMO was completely inactivated in 5 mM H2O2, mutant #16 retained >40% residual activity in 200 mM H2O2. In addition, the melting temperature of mutant #15 was increased by 7 °C compared to wild-type CHMOAcineto. | 87 |
Conclusions
In conclusion, this perspective article has shown that in recent years the number of Baeyer–Villiger monooxygenases useful for biocatalysis has substantially increased. Major reasons are novel tools to discover enzymes by protein sequence, phylogenetic and structural analysis or by identification of family relationships. This also helped to decipher possible natural functions of BVMOs and facilitated their improvement by protein engineering. Recently developed novel high-throughput assays will further contribute to identify or create novel BVMOs and to tailor-design their properties. Already, the larger “toolbox” of BVMOs available helped to substantially broaden their synthetic utility in organic chemistry. Furthermore, in the past decade, a range of factors limiting the application of BVMOs could be identified and tools to overcome these hurdles have been developed and already led to the first large scale applications of BVMOs. Overall, these achievements and efforts strongly helped to make BVMOs versatile enzymes for numerous applications in organic synthesis.We thank the Deutsche Forschungsgemeinschaft (Grant Bo1862/6-1), the Deutsche Bundesstiftung Umwelt (AZ13234) and the BMBF (Biokatalyse2021 cluster, FK0315175B) for financial support.
Notes and references
- H. Leisch, K. Morley and P. C. Lau, Chem. Rev., 2011, 111, 4165–4222 CrossRef CAS.
- D. E. Torres Pazmino, H. M. Dudek and M. W. Fraaije, Curr. Opin. Chem. Biol., 2010, 14, 138–144 CrossRef CAS.
- G. de Gonzalo, M. D. Mihovilovic and M. W. Fraaije, ChemBioChem, 2010, 11, 2208–2231 CrossRef CAS.
- J. Rehdorf and U. T. Bornscheuer, in Encyclopedia of Industrial Biotechnology. Bioprocess, Bioseparation and Cell Technology, ed. M. C. Flickinger, John Wiley & Sons, 2010, vol. 7 DOI:10.1002/9780470054581.eib451.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, K. Robins and J. C. Moore, Nature, 2012, 485, 185–194 CrossRef CAS.
- N. A. Donoghue, D. B. Norris and P. W. Trudgill, Eur. J. Biochem., 1976, 63, 175–192 CrossRef CAS.
- I. E. Conrad, R. Dubus, I. C. Gunsalus and N. York, Biochem. Biophys. Res. Commun., 1961, 6, 293–297 CrossRef.
- J. D. Stewart, Curr. Org. Chem., 1998, 2, 195–216 CAS.
- M. M. Kayser and C. M. Clouthier, J. Org. Chem., 2006, 71, 8424–8430 CrossRef CAS.
- K. H. Jones, R. T. Smith and P. W. Trudgill, J. Gen. Microbiol., 1993, 139, 797–805 CAS.
- H. Leisch, R. Shi, S. Grosse, K. Morley, H. Bergeron, M. Cygler, H. Iwaki, Y. Hasegawa and P. C. K. Lau, Appl. Environ. Microbiol., 2012, 78, 2200–2212 CrossRef CAS.
- M. Kadow, K. Loschinski, S. Sass, M. Schmidt and U. T. Bornscheuer, Appl. Microbiol. Biotechnol., 2012 DOI:10.1007/s00253-011-3859-1 , online.
- M. Kadow, S. Sass, M. Schmidt and U. T. Bornscheuer, AMB Express, 2011, 1, 13 CrossRef CAS.
- H. J. Ougham, D. G. Taylor and P. W. Trudgill, J. Bacteriol., 1983, 153, 140–152 CAS.
- M. W. Fraaije, N. M. Kamerbeek, W. J. van Berkel and D. B. Janssen, FEBS Lett., 2002, 518, 43–47 CrossRef CAS.
- M. W. Fraaije, J. Wu, D. P. H. M. Heuts, E. W. van Hellemond, J. H. L. Spelberg and D. B. Janssen, Appl. Microbiol. Biotechnol., 2005, 66, 393–400 CrossRef CAS.
- E. Malito, A. Alfieri, M. W. Fraaije and A. Mattevi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13157–13162 CrossRef CAS.
- A. Riebel, H. M. Dudek, G. de Gonzalo, P. Stepniak, L. Rychlewski and M. W. Fraaije, Appl. Microbiol. Biotechnol., 2012 DOI:10.1007/s00253-011-3823-0 , online.
- C. Szolkowy, L. D. Eltis, N. C. Bruce and G. Grogan, ChemBioChem, 2009, 10, 1208–1217 CrossRef CAS.
- H. Iwaki, S. Wang, S. Grosse, H. Bergeron, A. Nagahashi, J. Lertvorachon, J. Yang, Y. Konishi, Y. Hasegawa and P. C. Lau, Appl. Environ. Microbiol., 2006, 72, 2707–2720 CrossRef CAS.
- E. Beneventi, G. Ottolina, G. Carrea, W. Panzeri, G. Fronza and P. C. K. Lau, J. Mol. Catal. B: Enzym., 2009, 58, 164–168 CrossRef CAS.
- M. J. Fink, T. C. Fischer, F. Rudroff, H. Dudek, M. W. Fraaije and M. D. Mihovilovic, J. Mol. Catal. B: Enzym., 2011, 73, 9–16 CrossRef CAS.
- F. Leipold, R. Wardenga and U. T. Bornscheuer, Appl. Microbiol. Biotechnol., 2012, 94, 705–717 CrossRef CAS.
- J. B. van Beilen, F. Mourlane, M. A. Seeger, J. Kovac, Z. Li, T. H. Smits, U. Fritsche and B. Witholt, Environ. Microbiol., 2003, 5, 174–182 CrossRef CAS.
- D. G. Taylor and P. W. Trudgill, J. Bacteriol., 1986, 165, 489–497 CAS.
- R. Villa, J. Mol. Catal. B: Enzym., 1997, 2, 193–197 CrossRef CAS.
- M. J. van der Werf, H. J. Swarts and J. A. de Bont, Appl. Environ. Microbiol., 1999, 65, 2092–2102 CAS.
- E. J. McGhie, M. N. Isupov, E. Schroder and J. A. Littlechild, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1998, 54, 1035–1038 CrossRef CAS.
- J. Fried, R. W. Thoma and A. Klingsberg, J. Am. Chem. Soc., 1953, 75, 5764–5765 CrossRef CAS.
- A. Świzdor, T. Kołek, A. Panek and A. Białońska, Biochim. Biophys. Acta – Mol. Cell. Biol. L., 2011, 1811, 253–262 Search PubMed.
- G. J. Grogan and H. L. Holland, J. Mol. Catal. B: Enzym., 2000, 9, 1–32 CrossRef CAS.
- Z. Xiong, Q. Wei, H. Chen, S. Chen, W. Xu, G. Qiu, S. Liang and X. Hu, Steroids, 2006, 71, 979–983 CrossRef CAS.
- E. Itagaki, J. Biochem., 1986, 99, 825–832 CAS.
- K. Königsberger, G. Braunegg, K. Faber and H. Griengl, Biotechnol. Lett., 1990, 12, 509–514 CrossRef.
- P. C. Brzostowicz, D. M. Walters, S. M. Thomas, V. Nagarajan and P. E. Rouviere, Appl. Environ. Microbiol., 2003, 69, 334–342 CrossRef CAS.
- Y. C. Chen, O. P. Peoples and C. T. Walsh, J. Bacteriol., 1988, 170, 781–789 CAS.
- W. G. Lai, N. Farah, G. A. Moniz and Y. N. Wong, Drug Metab. Dispos., 2010, 39, 61–70 CrossRef.
- A. Rioz-Martínez, M. Kopacz, G. de Gonzalo, D. E. Torres Pazmiño, V. Gotor and M. W. Fraaije, Org. Biomol. Chem., 2011, 9, 1337–1341 Search PubMed.
- C. N. Jensen, J. Cartwright, J. Ward, S. Hart, J. P. Turkenburg, S. T. Ali, M. J. Allen and G. Grogan, ChemBioChem, 2012, 13, 872–878 CrossRef CAS.
- Y. Wen, H. Hatabayashi, H. Arai, H. K. Kitamoto and K. Yabe, Appl. Environ. Microbiol., 2005, 71, 3192–3198 CrossRef CAS.
- J. Jiang, C. N. Tetzlaff, S. Takamatsu, M. Iwatsuki, M. Komatsu, H. Ikeda and D. E. Cane, Biochemistry, 2009, 48, 6431–6440 CrossRef CAS.
- M.-J. Seo, D. Zhu, S. Endo, H. Ikeda and D. E. Cane, Biochemistry, 2011, 50, 1739–1754 CrossRef CAS.
- K. Qiao, Y.-H. Chooi and Y. Tang, Metab. Eng., 2011, 13, 723–732 CrossRef CAS.
- A. Kirschner, J. Altenbuchner and U. T. Bornscheuer, Appl. Microbiol. Biotechnol., 2007, 75, 1095–1101 CrossRef CAS.
- J. Rehdorf, A. Kirschner and U. T. Bornscheuer, Biotechnol. Lett., 2007, 29, 1393–1398 CrossRef CAS.
- J. Rehdorf, A. Lengar, U. T. Bornscheuer and M. D. Mihovilovic, Bioorg. Med. Chem. Lett., 2009, 19, 3739–3743 CrossRef CAS.
- J. Rehdorf, M. D. Mihovilovic, M. W. Fraaije and U. T. Bornscheuer, Chem.–Eur. J., 2010, 16, 9525–9535 CrossRef CAS.
- M. J. Fink, F. Rudroff and M. D. Mihovilovic, Bioorg. Med. Chem. Lett., 2011, 21, 6135–6138 CrossRef CAS.
- A. Rioz-Martínez, A. Cuetos, C. Rodríguez, G. de Gonzalo, I. Lavandera, M. W. Fraaije and V. Gotor, Angew. Chem., Int. Ed., 2011, 50, 8387–8390 CrossRef.
- P. B. Brondani, G. de Gonzalo, M. W. Fraaije and L. H. Andrade, Adv. Synth. Catal., 2011, 353, 2169–2173 CrossRef CAS.
- B. P. Branchaud and C. T. Walsh, J. Am. Chem. Soc., 1985, 107, 2153–2161 CrossRef CAS.
- S. Colonna, N. Gaggero, G. Carrea, G. Ottolina, P. Pasta and F. Zambianchi, Tetrahedron Lett., 2002, 43, 1797–1799 CrossRef CAS.
- L. H. Andrade, E. C. Pedrozo, H. G. Leite and P. B. Brondani, J. Mol. Catal. B: Enzym., 2011, 73, 63–66 CrossRef CAS.
- J. Zhu, Y. K. Bong, S. J. Collier, M. Vogel, M. J. Nazor, D. Smith, S. Song, M. D. Clay, B. Mijts and X. Zhang, 2011, Int. Patent Application to Codexis Inc., WO/2011/071982.
- H. E. M. Law, C. V. F. Baldwin, B. H. Chen and J. M. Woodley, Chem. Eng. Sci., 2006, 61, 6646–6652 CrossRef CAS.
- F. Secundo, S. Fialà, M. W. Fraaije, G. de Gonzalo, M. Meli, F. Zambianchi and G. Ottolina, Biotechnol. Bioeng., 2011, 108, 491–499 CrossRef CAS.
- G. de Gonzalo, C. Rodríguez, A. Rioz-Martínez and V. Gotor, Enzyme Microb. Technol., 2012, 50, 43–49 CrossRef CAS.
- F. Schulz, F. Leca, F. Hollmann and M. T. Reetz, Beilstein J. Org. Chem., 2005, 1, 10, DOI:10.1186/1860-5397-1-10.
- C. Rodríguez, G. de Gonzalo, M. W. Fraaije and V. Gotor, Green Chem., 2010, 12, 2255–2260 RSC.
- C. Rodríguez, G. de Gonzalo and V. Gotor, J. Mol. Catal. B: Enzym., 2012, 74, 138–143 CrossRef.
- A. Kheirolomoom, M. Ardjmand, M. Vossoughi and M. Kazemeini, Biochem. Eng. J., 1998, 2, 81–88 CrossRef CAS.
- C.-H. Wong, D. G. Drueckhammer and H. M. Sweers, J. Am. Chem. Soc., 1985, 107, 4028–4031 CrossRef CAS.
- C.-H. Wong and G. M. Whitesides, J. Am. Chem. Soc., 1981, 103, 4890–4899 CrossRef CAS.
- F. Zambianchi, P. Pasta, G. Carrea, S. Colonna, N. Gaggero and J. M. Woodley, Biotechnol. Bioeng., 2002, 78, 489–496 CrossRef CAS.
- J. M. Vrtis, A. K. White, W. W. Metcalf and W. A. van der Donk, Angew. Chem., Int. Ed., 2002, 41, 3257–3259 CrossRef CAS.
- R. Woodyer, W. A. van der Donk and H. Zhao, Biochemistry, 2003, 42, 11604–11614 CrossRef CAS.
- D. E. Torres Pazmino, R. Snajdrova, B. J. Baas, M. Ghobrial, M. D. Mihovilovic and M. W. Fraaije, Angew. Chem., Int. Ed., 2008, 47, 2275–2278 CrossRef.
- T. W. Johannes, R. D. Woodyer and H. Zhao, Biotechnol. Bioeng., 2007, 96, 18–26 CrossRef CAS.
- V. Alphand, G. Carrea, R. Wohlgemuth, R. Furstoss and J. M. Woodley, Trends Biotechnol., 2003, 21, 318–323 CrossRef CAS.
- S. D. Doig, H. Simpson, V. Alphand, R. Furstoss and J. M. Woodley, Enzyme Microb. Technol., 2003, 32, 347–355 CrossRef CAS.
- C. V. F. Baldwin, R. Wohlgemuth and J. M. Woodley, Org. Process Res. Dev., 2008, 12, 660–665 CrossRef CAS.
- S. D. Doig, P. J. Avenell, P. A. Bird, P. Gallati, K. S. Lander, G. J. Lye, R. Wohlgemuth and J. M. Woodley, Biotechnol. Prog., 2002, 18, 1039–1046 CrossRef CAS.
- H. D. Simpson, V. Alphand and R. Furstoss, J. Mol. Catal. B: Enzym., 2001, 16, 101–108 CrossRef CAS.
- I. Hilker, M. C. Gutierrez, R. Furstoss, J. Ward, R. Wohlgemuth and V. Alphand, Nat. Protoc., 2008, 3, 546–554 CrossRef CAS.
- I. Hilker, V. Alphand, R. Wohlgemuth and R. Furstoss, Adv. Synth. Catal., 2004, 346, 203–214 CrossRef CAS.
- B. H. Chen, S. D. Doig, G. J. Lye and J. M. Woodley, Trans. Inst. Chem. Eng., 2002, 80, 51–55 CAS.
- J. Rehdorf, C. L. Zimmer and U. T. Bornscheuer, Appl. Environ. Microbiol., 2009, 75, 3106–3114 CrossRef CAS.
- K. Geitner, J. Rehdorf, R. Snajdrova and U. T. Bornscheuer, Appl. Microbiol. Biotechnol., 2010, 88, 1087–1093 CrossRef CAS.
- O. Abril, C. C. Ryerson, C. T. Walsh and G. M. Whitesides, Bioorg. Chem., 1989, 17, 41–52 CrossRef CAS.
- K. S. Atia, Radiat. Phys. Chem., 2005, 73, 91–99 CrossRef CAS.
- A. Cuetos, A. Rioz-Martínez, M. L. Valenzuela, I. Lavandera, G. de Gonzalo, G. A. Carriedo and V. Gotor, J. Mol. Catal. B: Enzym., 2012, 74, 178–183 CrossRef CAS.
- S. A. Meeuwissen, A. Rioz-Martínez, G. de Gonzalo, M. W. Fraaije, V. Gotor and J. C. M. van Hest, J. Mater. Chem., 2011, 21, 18923 RSC.
- M. Hucik, M. Bucko, P. Gemeiner, V. Stefuca, A. Vikartovska, M. D. Mihovilovic, F. Rudroff, N. Iqbal, D. Chorvat, Jr. and I. Lacik, Biotechnol. Lett., 2010, 32, 675–680 CrossRef CAS.
- E.-H. Doo, W.-H. Lee, H.-S. Seo, J.-H. Seo and J.-B. Park, J. Biotechnol., 2009, 142, 164–169 CrossRef CAS.
- H. M. Kim, E. Heinzle and C. Wittmann, J. Microbiol. Biotechnol., 2006, 16, 1174–1179 CAS.
- J. Y. Yun, J. E. Lee, K. M. Yang, S. Cho, A. Kim, Y. E. Kwon and J. B. Park, Bioprocess Biosyst. Eng., 2012, 35, 211–216 CrossRef CAS.
- D. J. Opperman and M. T. Reetz, ChemBioChem, 2010, 11, 2589–2596 CrossRef CAS.
- R. Orru, H. M. Dudek, C. Martinoli, D. E. Torres Pazmino, A. Royant, M. Weik, M. W. Fraaije and A. Mattevi, J. Biol. Chem., 2011, 286, 29284–29291 CrossRef CAS.
- I. A. Mirza, B. J. Yachnin, S. Wang, S. Grosse, H. Bergeron, A. Imura, H. Iwaki, Y. Hasegawa, P. C. Lau and A. M. Berghuis, J. Am. Chem. Soc., 2009, 131, 8848–8854 CrossRef CAS.
- M. P. Beam, M. A. Bosserman, N. Noinaj, M. Wehenkel and J. Rohr, Biochemistry, 2009, 48, 4476–4487 CrossRef CAS.
- M. N. Isupov and A. A. Lebedev, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2008, 64, 90–98 CrossRef.
- A. B. Watts, J. Beecher, C. S. Whitcher and J. A. Littlechild, Biocatal. Biotransform., 2002, 20, 209–214 CrossRef CAS.
- M. C. Gutierrez, A. Sleegers, H. D. Simpson, V. Alphand and R. Furstoss, Org. Biomol. Chem., 2003, 1, 3500–3506 CAS.
- R. Sicard, L. Ä. Chen, A. Ä. Marsaioli and J.-L. Reymond, Adv. Synth. Catal., 2005, 347, 1041–1050 CrossRef CAS.
- D. Wahler and J. L. Reymond, Angew. Chem., Int. Ed., 2002, 41, 1229–1232 CrossRef CAS.
- A. Kirschner and U. T. Bornscheuer, Appl. Microbiol. Biotechnol., 2008, 81, 465–472 CrossRef CAS.
- J. A. Linares-Pastén, G. Chávez-Lizárraga, R. Villagomez, G. Mamo and R. Hatti-Kaul, Enzyme Microb. Technol., 2012, 50, 101–106 CrossRef.
- S. Saß, M. Kadow, K. Geitner, M. L. Thompson, L. Talmann, D. Böttcher, M. Schmidt and U. T. Bornscheuer, Tetrahedron, 2012 DOI:10.1016/j.tet.2012.05.098 , online.
- G. Behrens, A. Hummel, S. K. Padhi, S. Schätzle and U. T. Bornscheuer, Adv. Synth. Catal., 2011, 353, 2191–2215 CrossRef CAS.
- M. T. Reetz and G. P. L. Krebs, C. R. Chim., 2011, 14, 811–818 CrossRef CAS.
- Engineering Handbook, ed. S. Lutz and U. T. Bornscheuer, Wiley-VCH, 2009 Search PubMed.
- M. T. Reetz and S. Wu, Chem. Commun., 2008, 5499–5501 RSC.
- M. T. Reetz and S. Wu, J. Am. Chem. Soc., 2009, 131, 15424–15432 CrossRef CAS.
- S. Wu, J. P. Acevedo and M. T. Reetz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2775–2780 CrossRef CAS.
- H. M. Dudek, D. E. Torres Pazmino, C. Rodriguez, G. de Gonzalo, V. Gotor and M. W. Fraaije, Appl. Microbiol. Biotechnol., 2010, 88, 1135–1143 CrossRef CAS.
- H. M. Dudek, G. de Gonzalo, D. E. Torres Pazmino, P. Stepniak, L. S. Wyrwicz, L. Rychlewski and M. W. Fraaije, Appl. Environ. Microbiol., 2011, 77, 5730–5738 CrossRef CAS.
- H. L. van Beek, G. d. Gonzalo and M. W. Fraaije, Chem. Commun., 2012, 48, 3288 RSC.
- M. Bučko, A. Schenkmayerová, P. Gemeiner, A. Vikartovská, M. D. Mihovilovič and I. Lacík, Enzyme Microb. Technol., 2011, 49, 284–288 CrossRef.
Footnote |
| † All authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2012 |