Synthesis of anti-HIV lithospermic acid by two diverse strategies†
Tirumala G.
Varadaraju
a and
Jih Ru
Hwu
*ab
aDepartment of Chemistry and Frontier Research Center on Fundamental and Applied Sciences of Matters, National Tsing Hua University, Hsinchu, Taiwan 30013, R.O.C.
bDepartment of Chemistry, National Central University, Jhongli, Taoyuan, Taiwan 32001, R.O.C. E-mail: jrhwu@mx.nthu.edu.tw
First published on 14th May 2012
Abstract
An efficient and convergent route for the synthesis of the natural product (+)-lithospermic acid, which possesses anti-HIV activity, was accomplished. The (±)-trans-dihydrobenzo[b]furan core therein was prepared by two different strategies. The first strategy involved the use of a palladium-catalyzed annulation to generate an appropriately substituted benzo[b]furan ester followed by a stereoselective reduction of a carbon–carbon double bond with Mg–HgCl2–MeOH. The second strategy relied on an aldol condensation between a suitably substituted methyl arylacetate and 3,4-dimethoxybenzaldehyde, followed by cyclization. Finally, a total synthesis of (+)-lithospermic acid was completed via coupling of a trans-dihydrobenzo[b]furan cinnamic acid with an enantiomerically pure methyl lactate.
Introduction
In 1963, Johnson and co-workers1 reported the first isolation of lithospermic acid (1) from the roots of Lithospermum ruderale. In 1975, Carmack and co-workers2 isolated the same acid, 1, from the Lithospermum ruderale Dougl. ex Lehm (Boraginaceae). They proposed a new structure through the possession of a trans relationship between the C(20)/C(21) substituents attached to the dihydrobenzo[b]furan moiety, on the basis of 1H NMR coupling constants associated with similar compounds.2 In the same year, Wagner and co-workers3 isolated the same compound from Lithospermum officinale and confirmed the structure of 1. Moreover, they prepared its derivative (+)-heptamethyllithospermate. Though the structure of (+)-lithospermic acid (1) has been established independently by the groups of Carmack and Wagner, the absolute configuration remained unassigned.In 2002, Huang and co-workers4 reported their isolation of (+)-lithospermic acid (1) from Salvia miltiorrhiza and its non-toxic HIV-1 integrase inhibitory activity. In addition, this caffeic acid trimer (1)5 was found to possess inhibition properties of adenylatecyclase by Kohda et al.6 and an antioxidizing low-density lipoprotein by Lin et al.7

Given these remarkable biological properties, the total synthesis of (+)-lithospermic acid (1) and its analogues has attracted much attention. In 1979, Jacobson and Raths8 completed the synthesis of racemic heptamethyllithospermate. After two decades, Bergman, Ellman and co-workers9 accomplished the first asymmetric total synthesis of (+)-lithospermic acid (1) and established its absolute configuration as (+)-(10R,20S,21S)-1. Very recently, Wang and Yu10 have also reported a total synthesis of (+)-lithospermic acid. Meanwhile, Coster and co-workers11 disclosed the formal total synthesis of (+)-lithospermic acid (1) by using a late stage separation of the diastereomeric mixture. In these published works, Bergman, Ellman and Yu utilized naturally-occurring (R)-rosmarinic acid to generate the C(10R) stereogenic center in the natural product 1. To the best of our knowledge, no general synthetic route to establish the C(10R) chiral center from achiral starting material has been reported to date. Here, we report the first asymmetric synthesis of (+)-(R)-methyl 3-(3,4-dimethoxyphenyl)-2-hydroxypropanoate (3) as well as the establishment of the C(10R) stereogenic center of 1 by a chemical method. In addition, we demonstrate two synthetic sequences leading to the trans-dihydrobenzo[b]furan core of the natural product 1 from different starting materials. Furthermore, our success on the synthesis of (+)-lithospermic acid (1) via the intermediate 2 is described.
A retrosynthetic plan for (+)-lithospermic acid (1) is depicted in Scheme 1. We decided to synthesize the enantiomerically-enriched heptamethyllithospermate 2, in which demethylation of all seven methyl groups to give lithospermic acid had already been reported.9 Esterification of cinnamic acid 5 with (+)-(R)-methyl lactate 3 would give the intermediate 2. The (+)-lactate 3 could be synthesized from commercially available 3,4-dimethoxybenzaldehyde (4). Moreover, we planned to obtain the desired trans-dihydrobenzo[b]furan cinnamic acid 5 through two different strategies: namely palladium-catalyzed annulation and an aldol condensation.
 | ||
| Scheme 1 Retrosynthetic analysis of (+)-lithospermic acid (1) via its heptamethyl derivative 2. | ||
Results
At the outset, our approach towards obtaining the optically active lactate (R)-3 was to reduce the enol ester 9 (see Scheme 2). It can be prepared from 3,4-dimethoxybenzaldehyde (4) and N-acetylglycine in three steps by the Dalla's procedure.12 Although we were able to reduce 9 with NaBH4 to give the racemic lactate 3, use of chiral reducing agents including NB-Enantride, K-Glucoride and (R,R)-(−)-trans-4,5-bis[(diphenylphosphino)methyl]-2,2-di-methyl-1,3-dioxolane (i.e., DIOP–Rh(I)) did not lead to (+)-3 with a high % ee. Alternatively, we dehydroxylated the optically active diol 1013 selectively at the benzylic position14 by Et3SiH in the presence of CF3COOH at 0 °C. The desired (+)-methyl lactate 3 was generated in a 75% yield with a >99% ee. These mild conditions did not cause epimerization of the hydroxyl group at the α position. Its enantiomeric excess was determined by HPLC analysis with a Chiralcel OD column. Bergman, Ellman and co-workers9 reported a method for generating this optically active lactate (R)-3 by pentamethylation of commercially available rosmarinic acid followed by saponification. On the other hand, Eicher et al.15 obtained (+)-3 from its racemates by kinetic resolution with the enzyme Lipase PS. In comparison with their results, we successfully developed a simple approach for the enantioselective synthesis of (+)-(R)-3 and it can be regarded as a general route for the syntheses of other lactates.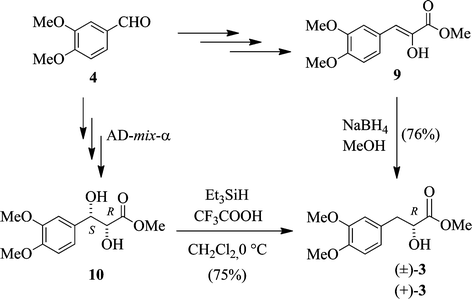 | ||
| Scheme 2 Syntheses of racemic and optically active (+)-(R)-methyl lactate 3. | ||
Next, our goal was to synthesize the key intermediate trans-dihydrobenzo[b]furan cinnamic acid 5. Recently, our group has utilized palladium-catalyzed annulation16 in the syntheses of functionalized benzo[b]furans and indoles for organic light-emitting diodes.17 We envisioned that similar chemistry could be applied to the synthesis of the dihydrobenzo[b]furan segment of (+)-lithospermic acid (1). Accordingly, we coupled 2-iodoisovanillin (6)18 with the activated alkyne 79 by using (PPh3)2PdCl2 (0.060 equiv) with LiCl and NaHCO3 in DMF at 110 °C to give the benzo[b]furan ester 11 as pale yellow needles (mp 166–167 °C, see Scheme 3). Subsequently, the chemo- and stereoselective reduction of 11 to the corresponding trans-dihydrobenzo[b]furan 13 was attempted. Application of the reduction conditions, including H2/Pd–C19 and Et3SiH/TFA,20 led to the recovery of the starting material or undesired products.
 | ||
| Scheme 3 A palladium-catalyzed annulation strategy in the synthesis of (±)-trans-dihydrobenzofuran cinnamic acid 5. | ||
In order to circumvent this problem, we considered metallic Mg in methanol (Mg–MeOH)21 as an ideal reducing system, which has been used in the reduction of α,β-unsaturated esters.21 Accordingly, benzo[b]furan ester 11 was produced up to a 30% yield (see entry 1 of Table 1). Furthermore, we treated compound 11 with Mg (40.0 equiv) in the presence of a catalytic amount of HgCl2 (0.10 equiv) by following the procedure reported by Lee et al. (entry 5 of Table 1).21a (±)-trans-Dihydrobenzo[b]furan 12 was generated as a pale yellow liquid in a 82% yield. Separation of this racemate to give the desired compound 12 in its enantiomerically pure form met with failure when a reported procedure was used.22 This racemic compound exhibited four singlets between 3.69 and 3.84 ppm in its 1H NMR spectrum for the 12 protons resulting from three unequivalent methoxy groups and one methyl ester. In addition, we observed two characteristic doublets with J = 6.0 Hz at 4.24 and 5.93 ppm for the C(3)- and C(2)-protons of dihydrobenzo[b]furan moiety, respectively, as well as a doublet at 4.49 ppm with J = 12.4 Hz for the C(8)H2 protons attached to a free hydroxyl group. Furthermore, in its 13C NMR spectrum, the resonance occurred at 55.94 and 87.08 ppm for the C(3)- and the C(2)-carbons, respectively, as well as at 62.66 ppm for the C(8) carbon. In its IR spectrum, one strong stretching absorption band appeared at 1736 cm−1 for the O(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) group and at 3516 cm−1 for the OH group. Moreover, its exact mass was detected as 374.1366, which agreed well with the theoretical value of 374.1360 for C20H22O7. These spectroscopic data clearly confirm the structure of trans-dihydrobenzo[b]furan 12.
O) group and at 3516 cm−1 for the OH group. Moreover, its exact mass was detected as 374.1366, which agreed well with the theoretical value of 374.1360 for C20H22O7. These spectroscopic data clearly confirm the structure of trans-dihydrobenzo[b]furan 12.
| Entry | Mg (equiv) | HgCl2 (equiv) | Temp (°C) | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| 1 | 40.0 | — | 25 | 48 | 30 |
| 2 | 40.0 | — | 65 | 12 | 0 |
| 3 | 5.0 | 0.10 | 25 | 3.0 | 35 |
| 4 | 40.0 | 0.10 | 25 | 3.0 | 60 |
| 5 | 40.0 | 0.10 | 25 | 20 | 82 |
We oxidized alcohol 12 to the corresponding aldehyde 13,8,9 known to possess a trans configuration, by manganese dioxide.23 Finally, the Knoevenagel condensation of aldehyde (±)-13 with malonic acid under basic conditions was performed by use of the Ellman's procedure to give the desired (±)-trans-dihydrobenzo[b]furan cinnamic acid 5.8,9
At this stage, we planned a completely different approach for the synthesis of dihydrobenzo[b]furan 5, commencing with o-vanillin (14). This was converted to 2-benzyloxy-6-bromo-3-methoxybenzaldehyde (15) in four steps by the procedure reported by Nakanishi (Scheme 4).24 Condensation of aldehyde 15 with CH3SCH2SOCH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25 in the presence of benzyltrimethylammonium hydroxide (Triton B) at 100 °C gave the ketenethioacetal 16. Upon acid-catalyzed hydrolysis in dry methanol, the desired methyl arylacetate 8 was formed in an 86% yield. We then performed an aldol condensation26 between 8 and 3,4-dimethoxybenzaldehyde (4) in the presence of lithium diisopropylamide (LDA) at −78 °C to exclusively give a coupled (±)-β-hydroxy ester as white solids (mp 95–96 °C) in a 65% yield . At this time, the relative stereochemistry of the product was not important and was tentatively assigned as 17. The hydroxyl group therein could be removed to form a carbocationic intermediate for cyclization, after which the required trans configuration of the dihydrofuran product must be ensured.27 Subsequent cyclization of compound 17 with iodotrimethylsilane (2.2 equiv)28 in dichloromethane led to the desired bromodihydrobenzo[b]furan 18 as a gummy oil in a 75% yield.
25 in the presence of benzyltrimethylammonium hydroxide (Triton B) at 100 °C gave the ketenethioacetal 16. Upon acid-catalyzed hydrolysis in dry methanol, the desired methyl arylacetate 8 was formed in an 86% yield. We then performed an aldol condensation26 between 8 and 3,4-dimethoxybenzaldehyde (4) in the presence of lithium diisopropylamide (LDA) at −78 °C to exclusively give a coupled (±)-β-hydroxy ester as white solids (mp 95–96 °C) in a 65% yield . At this time, the relative stereochemistry of the product was not important and was tentatively assigned as 17. The hydroxyl group therein could be removed to form a carbocationic intermediate for cyclization, after which the required trans configuration of the dihydrofuran product must be ensured.27 Subsequent cyclization of compound 17 with iodotrimethylsilane (2.2 equiv)28 in dichloromethane led to the desired bromodihydrobenzo[b]furan 18 as a gummy oil in a 75% yield.
![An alternative strategy to construct the dihydrobenzo[b]furan cinnamic acid 5 by use of an aldol condensation.](/image/article/2012/OB/c2ob25575h/c2ob25575h-s4.gif) | ||
| Scheme 4 An alternative strategy to construct the dihydrobenzo[b]furan cinnamic acid 5 by use of an aldol condensation. | ||
In the IR spectrum of the new dihydrobenzo[b]furan 18, strong absorptions resulting from stretching vibrations appeared at 1736 cm−1 for the ester CO group. Its 1H NMR spectrum displayed two doublets at 5.82 and 4.30 ppm with J = 7.2 Hz for the H(2) and H(3) protons, respectively, and one singlet at 3.73 ppm for the CO2CH3 protons. Although the JH(2)–H(3) value is not diagnostic to distinguish the cis and trans isomers, the anisotropic effect of the C(2) aryl group causes a chemical shift of the H(3) proton upfield and the CO2CH3 protons downfield in the trans isomers than those of the corresponding cis isomers.11 The trans compounds often display the H(3) protons around 4.3 ppm and the CO2CH3 protons around 3.8–3.9 ppm, yet the cis compounds display the H(3) protons around 4.9 ppm and the CO2CH3 protons around 3.3 ppm. Our experimental data clearly indicate the trans configuration of the dihydrobenzofuran moiety in compound 18.
Afterwards, we performed a Heck coupling29 between furan 18 and tert-butyl ester, using Pd(OAc)2 (0.10 equiv) and PPh3 (0.30 equiv) in Et3N, to produce an α,β-unsaturated tert-butyl ester 19 in 81% yield. Finally, treatment of ester 19 with trifluoroacetic acid gave the desired (±)-trans-dihydrobenzo[b]furancinnamic acid 5 as a pale yellow solid in a 92% yield.8,9 The physical properties and spectroscopic characteristics obtained by us for compound 5 are consistent with the reported data.8,9
Finally, to obtain heptamethyllithospermate 2, we esterified9 (±)-5 with (+)-R-methyl lactate 3 in the presence of 4-dimethylaminopyridine (DMAP), 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide (EDCI) and 1-hydroxybenzotriazole (HOBt, Scheme 4). A 1:1 mixture of (10R, 20S, 21S)-2 and the corresponding diastereomeric counterpart (10R,20R,21R)-2 was produced as a yellow foam in a 85% overall yield. Their physical properties and spectroscopic characteristics are consistent with those of the reported compounds.8,11 For example, a new characteristic peak at δ 5.26–5.35 ppm in the 1H NMR spectrum for the proton in –O(CO)CHCOO– indicates the formation of a new ester bond. Further conversion of (10R,20S,21S)-2 to the final target (+)-1 through multi-demethylation has been reported by Coster,11 Bergman and Ellman.9
Discussion
Antus,19 Rupprecht,30 and their co-workers reported that hydrogenation of 2,3-disubstituted benzo[b]furans with hydrogen gas in the presence of 10% palladium on carbon (Pd/C) affords the corresponding 2,3-dihydrobenzo[b]furans with a cis configuration. Isomerization of this cis isomer occurs under alkaline conditions at an elevated temperature to give the thermodynamically more stable trans diastereomer.30 Using this method, we hydrogenated benzo[b]furan ester 11 in methanol19 for 3 h. 4-(Hydroxymethyl)benzo[b]furan 20 was obtained, in which the C(2)C(3) double bond remained intact (see Scheme 5). Elongation of the hydrogenation time to 24 h produced 4-methylbenzo[b]furan 21 through deoxygenation of the benzyl alcohol moiety in 20. Our results reveal that the formyl group in 11 was problematic and converted to an undesired methyl group before the conjugated C(2)C(3) bond in the benzo[b]furan nucleus was hydrogenated.
![Hydrogenation of benzo[b]furan ester 11 containing a formyl group.](/image/article/2012/OB/c2ob25575h/c2ob25575h-s5.gif) | ||
| Scheme 5 Hydrogenation of benzo[b]furan ester 11 containing a formyl group. | ||
On the other hand, we endeavoured to circumvent the difficulty of the diastereoselective reduction of 2,3-disubstitutedbenzo[b]furan by using metallic Mg in methanol.21 Accordingly, the single electron transfer took place in benzo[b]furan ester 11 to chemo- and stereoselectively afford the hydroxymethyl trans-dihydrobenzo[b]furan 12 in a 30% yield (see Scheme 3 and entry 1 of Table 1). The yield was improved to 82% by the addition of a catalytic amount of HgCl2. Lee21a reported that the use of HgCl2 can activate the Mg and thus accelerate the reduction. Moreover, generation of magnesium methoxide in situ was responsible for the base-promoted epimerization at the C(3) carbonyl ester to the trans-diastereomer 12. In this regard, metallic Mg in a protic solvent acted not only as a single electron transfer reducing reagent but also the source of a base for epimerization.21
During the preparation of this manuscript, we noted that Coster and co-workers11 obtained a similar key intermediate as (±)-trans-dihydrobenzo[b]furan 13 for the synthesis of lithospermic acid (1). It includes the Sonogashira coupling, protection of the C(4)-formyl group, Pd(II)-catalyzed carbonylative annulation, Mg-mediated reduction and finally deprotection. Through a different pathway, we were able to obtain the desired (±)-trans-13 in three steps, including the palladium-catalyzed annulation,16 Mg/HgCl2-mediated reduction, and oxidation with a 48% overall yield.
Conclusion
The first asymmetric syntheses of (+)-(R)-methyl lactate 3 (>99%ee) was completed in four steps with an overall yield of 75% from commercially available 3,4-dimethoxybenzaldehyde. Two different synthetic routes were also developed for the diastereoselective synthesis of the trans-dihydrobenzo[b]furan segment of lithospermic acid (1). The first strategy involved the chemo- and stereoselective reduction of benzo[b]furan ester 11 with metallic Mg and catalytic HgCl2 in methanol. The second strategy involved an aldol condensation and subsequent intramolecular cyclization of β-hydroxy ester 17 to give the desired trans-dihydrobenzo[b]furan core of compound 1. These two strategies demonstrate expedient and concise routes for the synthesis of heptamethyllithospermate 2 from readily available starting materials in five and seven steps, respectively. Moreover, these two strategies were applied successfully in a total synthesis of (+)-lithospermic acid (1).Experimental section
General methods
All reactions were carried out in oven-dried glassware (120 °C) under an atmosphere of nitrogen unless as indicated otherwise. Ethyl acetate and hexane from Mallinckrodt Chemical Co. were dried and distilled from CaH2. Diethyl ether and THF from Mallinckrodt Chemicals Co. were dried by distillation from sodium and benzophenone under an atmosphere of nitrogen. Acetonitrile, acetone, dichloromethane and methanol were purchased from Mallinckrodt Chemical Co. Acetic anhydride, tert-butanol and pyridine were purchased from Echo Chemical Co. AD-mix-α, benzyltrimethylammonium hydroxide (Triton B), tert-butyl acrylate, bromine, n-butyllithium, carbon tetrabromide (CBr4), 4-dimethylaminopyridine (DMAP), 2,2-dimethyl-1,3-propanediol, 3,4-dimethoxybenzaldehyde, p-dioxane, 1,2-ethanediol, 1-hydroxybenzotriazole (HOBt), iodine monochloride, iodotrimethylsilane, isovanillin, magnesium, manganese dioxide, mercuric chloride, methanesulfonamide, palladium acetate, potassium bromide, potassium iodide, p-toluenesulfonic acid (PTSA), triethylsilane, trifluoroacetic acid, triphenylphosphine, and o-vanillin were purchased from Aldrich Chemical Co. Benzyl bromide (BnBr), bis(triphenylphosphine)palladium(II)chloride, diisopropylamine, 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide hydrochloride (EDCI), methyl chloroformate, methyl (methylthio)methyl sulfoxide (CH3SCH2SOCH3) and 1,3-propanedithiol were purchased from Fluka Chemical Co. Methyl 2-oxo-3-(3,4-dimethoxyphenyl)propionate (9),12 (2R,3S)-methyl 3-(3,4-dimethoxyphenyl)-2,3-dihydroxypropanoate (10),13 4-formyl-2-hydroxy-3-iodoanisole (6),18 (3,4-dimethoxyphenyl)propynoic acid methyl ester (7)9 and 2-benzyloxy-6-bromo-3-methoxybenzaldehyde (15)24 were prepared by literature methods.Analytical thin layer chromatography (TLC) was performed on precoated plates (silica gel 60 F-254), which were purchased from Merck Inc. Purification by gravity column chromatography was carried out by use of Silicycle ultra pure silica gel (particle size 40–63 μm, 230–400 mesh). The enantiomeric excess was determined by an HPLC (Waters 515) with Chiralcel OD (250 × 4.6 mm) column at 25 °C. The results were compared with racemic isomers with a flow rate of 1.0 mL min−1 and with isopropyl alcohol in hexane as the mobile phase. Purity of all the products was >98.0%, as checked by an HPLC (Waters 515) with a Lichrosorb Si-100 (200 × 4.6 mm, 5 μm) column at 25 °C.
Infrared (IR) spectra were measured on a Perkin–Elmer model spectrum one B spectrophotometer. Absorption intensities are recorded by the following abbreviations: s = strong; m = medium and w = weak. Proton NMR spectra were obtained on a Varian Mercury-400 (400 MHz) spectrometer, using chloroform-d (CDCl3) and acetone-d6 (CD3COCD3) as solvents. Proton NMR chemical shifts were referenced to residual protonated solvents (δ 7.24 and 2.05 ppm for chloroform and acetone, respectively). Carbon-13 NMR spectra were obtained on a Varian Mercury-400 (100 MHz) spectrometer using chloroform-d as the solvent. Carbon-13 chemical shifts are referenced to the center of the CDCl3 triplet (δ 77.0 ppm). Multiplicities are recorded by the following abbreviations: s = singlet; d = doublet; t = triplet; q = quartet; m = multiplet; J = coupling constant (hertz). High-resolution mass spectra were obtained by means of a JEOL JMS-700 mass spectrometer. A Perkin-Elmer 241 polarimeter with a sodium lamp was used for the determination of specific rotations at room temperature. Melting points were obtained with a Fargo MP-2D melting point apparatus.
(+)-(R)-Methyl 3-(3,4-dimethoxyphenyl)-2-hydroxypropanoate ((+)-3)
To a solution containing (2R,3S)-methyl 3-(3,4-dimethoxyphenyl)-2,3-dihydroxypropanoate13 (10, 542.1 mg, 2.117 mmol, 1.0 equiv) and triethylsilane (270.4 mg, 2.326 mmol, 1.1 equiv) in CH2Cl2 (20 mL) at 0 °C, trifluoroacetic acid (3.671 g, 32.17 mmol, 15.0 equiv) was added via a syringe and stirring was continued for 30 min. The reaction was quenched with saturated aqueous NaHCO3 (20 mL) and the resultant solution was extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were washed with a saturated aqueous NaCl solution (20 mL), dried over MgSO4(s), filtered and concentrated under reduced pressure to afford a residue. Purification of the residue by chromatography with a silica gel column (40% EtOAc in hexane as the eluent) gave (+)-3 (381.1 mg, 1.587 mmol) as a white solid in a 75% yield with >99%ee, as determined by HPLC with a Chiralcel OD column and isopropyl alcohol (10%) in hexane as the eluent. The major fraction showed up at tr = 18.94 min and the minor fraction at tr = 16.02 min by use of a UV detector with λ = 279 nm. For (+)-3: mp (recrystallized from toluene) 56–57 °C, lit.9 (53–54 °C); [α]D23 +9.9° (c = 0.77 in CH2Cl2), lit.9 +10.6° (c = 0.67 in CH2Cl2); TLC Rf: 0.56 (60% EtOAc in hexane as the eluent); 1H NMR (CDCl3; 400 MHz) δ 2.92 (dd, J = 14.0, 5.6 Hz, 1 H, ArCH), 3.07 (dd, J = 14.0, 5.6 Hz, 1 H, ArCH), 3.76 (s, 3 H, CO2CH3), 3.83 (s, 3 H, OCH3), 3.84 (s, 3 H, OCH3), 4.40–4.43 (m, 1 H, CHO), 6.71–6.79 (m, 3 H, 3 × ArH); 13C NMR (CDCl3; 100 MHz) δ 39.91, 52.18, 55.62, 55.65, 71.24, 111.00, 112.54, 121.31, 128.65, 147.81, 148.59, 174.35 (CO). Its physical properties and spectroscopic characteristics are consistent with those previously reported.9 The corresponding racemate (±)-3 was prepared through an established method12 and its 1H NMR and 13C NMR characteristics are consistent with those of (+)-3.
Methyl 2-(3,4-dimethoxyphenyl)-4-formyl-7-methoxybenzo[b]furan-3-carboxylate (11)
To a solution containing 4-formyl-2-hydroxy-3-iodoanisole18 (6, 152.6 mg, 0.5488 mmol, 1.0 equiv), (Ph3P)2PdCl2 (23.11 mg, 0.0329 mmol, 0.060 equiv), LiCl (23.26 mg, 0.5488 mmol, 1.0 equiv) and NaHCO3 (230.5 mg, 2.744 mmol, 5.0 equiv) in DMF (20 mL) (3,4-dimethoxyphenyl)propynoic acid methyl ester9 (7, 145.0 mg, 0.6584 mmol, 1.2 equiv) was added. After the reaction mixture was stirred under nitrogen at 110 °C for 6 h, it was quenched with saturated aqueous NH4Cl (50 mL) and extracted with Et2O (3 × 10 mL). The combined organic layers were washed with a saturated aqueous NaCl solution (10 mL), dried over MgSO4(s), filtered and concentrated under reduced pressure to afford a residue. Purification of the residue by chromatography with a silica gel column (30% EtOAc in hexane as the eluent) gave 11 (125.9 mg, 0.3402 mmol) as pale yellow crystalline solids in a 62% yield: mp (recrystallized from EtOAc) 166–167 °C; TLC Rf 0.36 (40% EtOAc in hexane as the eluent); 1H NMR (CDCl3; 400 MHz) δ 3.92 (s, 3 H, CO2CH3), 3.95 (s, 3 H, OCH3), 3.97 (s, 3 H, OCH3), 4.10 (s, 3 H, OCH3), 6.92 (d, J = 8.4 Hz, 1 H, ArH), 6.94 (d, J = 8.4 Hz, 1 H, ArH), 7.49–7.54 (m, 2 H, 2 × ArH), 7.75 (d, J = 8.4 Hz, 1 H, ArH), 10.03 (s, 1 H, CHO); 13C NMR (CDCl3; 100 MHz) δ 52.44, 55.85, 55.90, 56.26, 106.19, 110.10, 110.42, 111.00, 120.94, 121.40, 122.88, 126.79, 131.64, 142.85, 148.86, 149.61, 150.69, 157.10, 166.18 (CO), 189.63 (CO); IR (KBr) 3010 (w), 2031 (w), 1722 (s, CO), 1677 (s), 1617 (m), 1575 (s), 1516 (s), 1499 (s), 1399 (m), 1300 (s), 803 cm−1 (m); MS (FAB) m/z 370 (M+, 75), 339 (100), 311 (9), 221 (15), 165 (8); HRMS (FAB) calcd for C20H18O7: 370.1053, found 370.1050.
(±)-Methyl trans-2-(3,4-dimethoxyphenyl)-4-hydroxymethyl-7-methoxy-2,3-dihydrobenzo[b]furan-3-carboxylate (12)
To a solution containing aldehyde 11 (150.1 mg, 0.4055 mmol, 1.0 equiv) and Mg (394.3 mg, 16.22 mmol, 40.0 equiv) in tetrahydrofuran (10 mL) and methanol (10 mL) HgCl2 (11.01 mg, 0.0405 mmol, 0.10 equiv) was added. After the reaction mixture was stirred at room temperature for 48 h, it was quenched with HCl(aq) (5.0 N, 20 mL) and extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were washed with a saturated aqueous NaCl solution (15 mL), dried over MgSO4(s), filtered and concentrated under reduced pressure to afford a residue. Purification of the residue by chromatography with a silica gel column (35% EtOAc in hexane as the eluent) gave (±)-12 (124.4 mg, 0.3325 mmol) as a pale yellow liquid in a 82% yield: TLC Rf 0.37 (40% EtOAc in hexane as the eluent); 1H NMR (CDCl3; 400 MHz) δ 3.74 (s, 3 H, OCH3), 3.79 (s, 3 H, OCH3), 3.79 (s, 3 H, OCH3), 3.84 (s, 3 H, CO2CH3), 4.24 (d, J = 6.0 Hz, 1 H, ArCHCO2), 4.49 (d, J = 12.4 Hz, 2 H, CH2O), 5.93 (d, J = 6.0 Hz, 1 H, ArOCHAr), 6.75–6.87 (m, 5 H, 5 × ArH); 13C NMR (CDCl3; 100 MHz) δ 52.82, 55.71, 55.76, 55.77, 55.94, 62.66, 87.08, 108.72, 110.96, 112.54, 118.01, 121.48, 121.49, 123.10, 130.20, 132.47, 144.12, 148.26, 149.00, 172.58 (CO); IR (neat) 3516 (br, OH), 2952 (m), 2834 (w), 1736 (s, CO), 1593 (m), 1515 (s), 1438 (s), 1160 (s), 1024 (s), 808 (m) cm−1; MS (FAB) m/z 374 (M+, 9), 297 (37), 154 (55), 136 (70), 55 (100); HRMS (FAB) calcd for C20H22O7: 374.1366, found 374.1360.
(±)-Methyl trans-2-(3,4-dimethoxyphenyl)-4-formyl-7-methoxy-2,3-dihydrobenzo[b]-furan-3-carboxylate (13)
To a solution containing alcohol (±)-12 (112.6 mg, 0.3009 mmol, 1.0 equiv) in dichloromethane (10 mL) manganese dioxide (130.7 mg, 1.504 mmol, 5.0 equiv) was added. After stirring at room temperature for 48 h, the reaction mixture was filtered through Celite and concentrated under reduced pressure to afford a residue. Purification of the residue by chromatography with a silica gel column (30% EtOAc in hexane as the eluent) gave (±)-13 (106.3 mg, 0.2856 mmol) as pale yellow crystals in a 95% yield: mp (recrystallized from EtOAc) 48–49 °C; TLC Rf 0.47 (40% EtOAc in hexane as the eluent); 1H NMR (CDCl3; 400 MHz) δ 3.75 (s, 3 H, OCH3), 3.83 (s, 6 H, 2 × OCH3), 3.96 (s, 3 H, CO2CH3), 4.69 (d, J = 7.0 Hz, 1 H, ArCHCO2), 5.85 (d, J = 7.0 Hz, 1 H, ArOCHAr), 6.82 (d, J = 8.0 Hz, 1 H, ArH), 6.90–6.97 (m, 3 H, 3 × ArH), 7.40 (d, J = 8.4 Hz, 1 H, ArH), 9.81 (s, 1 H, CHO); 13C NMR (CDCl3; 100 MHz) δ 52.57, 55.85, 55.86, 56.16, 56.52, 88.73, 108.71, 110.96, 111.60, 118.17, 124.49, 126.50, 128.52, 132.01, 149.13, 149.23, 149.47, 149.54, 171.82 (CO), 190.56 (CO); MS (EI) m/z (relative intensity) 372 (M+, 100), 313 (68), 272 (11), 155 (13), HRMS (EI) calcd for C20H20O7: 372.1209, found 372.1176. Its spectroscopic characteristics are consistent with those previously reported.8
(±)-(2E)-3-[2-(3,4-Dimethoxyphenyl)-7-methoxy-3-methoxycarbonyl-2,3-dihydro benzo[b]furan-4-yl]prop-2-enoic acid (5): method 1
To a solution containing aldehyde (±)-13 (51.20 mg, 0.1375 mmol, 1.0 equiv) and malonic acid (43.21 mg, 0.4152 mmol, 3.0 equiv) in pyridine (10 mL) piperidine (68.79 mg, 0.6875 mmol, 5.0 equiv) was added. After the reaction mixture was stirred under nitrogen at 100 °C for 8 h, it was quenched with saturated aqueous NH4Cl (50 mL) and extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with HCl(aq) (1.0 N, 30 mL), dried over MgSO4(s), filtered and concentrated under reduced pressure to afford a residue. Purification of the residue by chromatography with a silica gel column (85% EtOAc in hexane as the eluent) gave (±)-5 (48.20 mg, 0.1169 mmol) as a light yellow solid in a 85% yield: mp (recrystallized from EtOAc) 65–66 °C; TLC Rf 0.47 (40% EtOAc in hexane as the eluent); 1H NMR (CDCl3; 400 MHz) δ 3.81 (s, 3 H, OCH3), 3.87 (s, 6 H, 2 × OCH3), 3.93 (s, 3 H, CO2CH3), 4.52 (d, J = 5.6 Hz, 1 H, ArCHCO2), 6.07 (d, J = 5.6 Hz, 1 H, ArOCHAr), 6.31 (d, J = 15.8 Hz, 1 H, Ph–CCH–CO2), 6.85–6.92 (m, 4 H, 4 × ArH), 7.26 (d, J = 8.8 Hz, 1 H, ArH), 7.84 (d, J = 15.8 Hz, 1 H, Ph–CHC–CO2); 13C NMR (CDCl3; 100 MHz) δ 52.80, 55.81, 55.82, 55.83, 56.07, 87.36, 108.69, 111.09, 112.91, 116.61, 117.96, 120.77, 124.04, 125.14, 132.15, 143.12, 146.44, 148.36, 149.15, 149.21, 171.68 (CO), 172.20 (CO). Its spectroscopic characteristics are consistent with those previously reported.9,11
(±)-(2E)-3-[2-(3,4-Dimethoxyphenyl)-7-methoxy-3-methoxycarbonyl-2,3-dihydro benzo[b]furan-4-yl]prop-2-enoic Acid (5): method 2
To a solution containing tert-butyl ester(±)-19 (21.20 mg, 0.045 mmol, 1.0 equiv) in CH2Cl2 (15 mL) trifluoroacetic acid (5.41 mg, 0.4509 mmol, 10.0 equiv) was added via a syringe at 0 °C. After stirring at room temperature for 2 h, it was quenched with saturated aqueous NaHCO3 (5.0 mL) and the resultant solution was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with a saturated aqueous NaCl solution (10 mL), dried over MgSO4(s), filtered and concentrated under reduced pressure to afford a residue. Purification of the residue by chromatography with a silica gel column (90% EtOAc in hexane as the eluent) gave (±)-5 (17.14 mg, 0.0416 mmol) as a pale yellow solid in a 92% yield. Its spectroscopic characteristics are consistent with those previously reported.9(10R,20S,21S)-Methyl 2-(3,4-dimethoxyphenyl)-4-([3-(3,4-dimethoxyphenyl)-1-methoxy-1-oxopropan-2-yloxy]-3-oxoprop-1-enyl)-7-methoxy-2,3-dihydrobenzo[b]furan-3-carboxylate (2) and its (10R,20R,21R)-diastereomer (2)
To a solution containing cinnamic acid (±)-5 (55.21 mg, 0.1333 mmol, 1.0 equiv), EDCI (28.11 mg, 0.1466 mmol, 1.1 equiv), DMAP (17.92 mg, 0.1466 mmol, 1.1 equiv) and HOBt (19.80 mg, 0.1466 mmol, 1.1 equiv) in CHCl3 (10 mL) (+)-methyl lactate (3, 38.40 mg, 0.1596 mmol, 1.2 equiv) was added at 0 °C. After the reaction mixture was stirred under nitrogen at room temperature for 48 h, it was quenched with saturated aqueous NH4Cl (20 mL) and extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were washed with a saturated aqueous NaCl solution (15 mL), dried over MgSO4(s), filtered and concentrated under reduced pressure to afford a residue. Purification of the residue by chromatography with silica gel column (40% EtOAc in hexane as the eluent) gave a 1:1 mixture of the title compound (10R,20S,21S)-2 and the corresponding diastereomeric counterpart (10R,20R,21R)-2 (72.19 mg, 0.1135 mmol) as a yellow foam in a 85% overall yield: TLC Rf 0.43 (40% EtOAc in hexane as the eluent); 1H NMR (CDCl3; 400 MHz) δ 3.15–3.16 (m, 2 H, ArCH2), 3.70 (s, 6 H, 2 × CO2CH3), 3.71 (s, 3 H, OCH3), 3.80 (s, 6 H, 2 × OCH3), 3.81 (s, 3 H, OCH3), 3.82 (s, 6 H, 2 × OCH3), 3.91 (s, 3 H, OCH3), 4.40 (d, J = 5.6 Hz, 1 H, ArCHCO2), 5.26–5.35 (m, 1 H, OCHCOO), 5.99–6.00 (m, 1 H, ArOCHAr), 6.30 (dd, diastereomers, J = 16.0, 4.2 Hz, 1 H), 6.73–6.87 (m, 7 H, 7 × ArH), 7.19 (m, 1 H, ArH), 7.73 (dd, diastereomers, J = 16.0, 4.2 Hz, 1 H); 13C NMR (CDCl3; 100 MHz) δ 37.09, 37.12, 52.27, 52.78, 52.74, 55.78, 55.81, 55.82, 55.90, 55.92, 56.07, 56.14, 73.07, 87.41, 108.77, 108.79, 110.55, 111.13, 111.16, 112.44, 112.48, 112.93, 112.96, 116.37, 116.42, 117.98, 118.01, 120.54, 120.81, 121.40, 121.45, 124.25, 124.34, 124.95, 125.14, 128.30, 132.27, 132.30, 135.26, 142.28, 142.41, 146.35, 148.04, 148.06, 148.40, 148.45, 148.78, 149.23, 149.30, 166.03, 170.23, 171.62, 171.70. The spectral data are consistent with those previously reported.8,11
2-Benzyloxy-6-bromo-3-methoxy-2-[2-(methylsulfinyl)-2-(methylthio)vinyl] benzene (16)
To a solution containing aldehyde2415 (2.125 g, 6.617 mmol, 1.0 equiv) and Triton B (4.0 ml) in THF (10 mL) CH3SCH2SOCH3 (986.2 mg, 7.939 mmol, 1.2 equiv) was added via a syringe at 0 °C. After the reaction mixture was refluxed at 100 °C for 12 h, it was quenched with saturated aqueous NH4Cl (20 mL) and the resultant solution was extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were washed with a saturated aqueous NaCl solution (50 mL), dried over MgSO4(s), filtered and concentrated under reduced pressure to afford a residue. Purification of the residue by chromatography with a silica gel column (20% EtOAc in hexane as the eluent) gave 16 (2.260 g, 5.305 mmol) as a yellow gum in a 80% yield: TLC Rf 0.51 (25% EtOAc in hexane as the eluent); 1H NMR (CDCl3; 400 MHz) δ 2.11 (s, 3 H, SMe), 2.60 (s, 3 H, SOMe), 3.85 (s, 3 H, OCH3), 4.95 (dd, J = 11.2, 10.8 Hz, 2 H, PhCH2), 6.82 (d, J = 8.8 Hz, 1 H, ArH), 7.24–7.35 (m, 6 H, 6 × ArH), 7.44 (s, 1 H, CC–H); 13C NMR (CDCl3; 100 MHz) δ 16.75, 40.14, 55.63, 74.77, 113.01, 113.04, 127.32, 127.73, 127.74, 127.75, 127.82, 128.00, 129.95, 132.11, 136.66, 145.70, 146.66, 151.87; IR (neat) 3391 (w), 3003 (m), 2838 (m), 1567 (s), 1528 (s), 1436 (s), 1350 (s), 1293 (s), 1063 (s), 979 (s), 883 (m), 802 (s), 733 (s), 698 (s) cm−1; MS (FAB) m/z 427 (M+ + 2, 9), 425 (M+, 9), 347 (16), 287 (40), 147 (55), 91 (100); HRMS (FAB) calcd for C18H19BrO3S2: 425.9959, found 425.9956.
Methyl 2-(2-benzyloxy-6-bromo-3-methoxyphenyl)ethanoate (8)
To a solution containing ketenethioacetal 16 (1.256 g, 2.948 mmol, 1.0 equiv) in dry MeOH (25 mL) HCl (g) was added at room temperature for 2 h. After the reaction mixture was stirred for 2 h, the solvent therein was evaporated under reduced pressure and the resultant solution was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with a saturated aqueous NaCl solution (20 mL), dried over MgSO4(s), filtered and concentrated under reduced pressure to afford a residue. Purification of the residue by chromatography with a silica gel column (20% EtOAc in hexane as the eluent) gave 8 (927.1 mg, 2.546 mmol) as a yellow liquid in a 86% yield: TLC Rf 0.38 (30% EtOAc in hexane as the eluent); 1H NMR (CDCl3; 400 MHz) δ 3.62 (s, 3 H, OCH3), 3.82 (s, 2 H, CH2CO2), 3.85 (s, 3 H, CO2CH3), 5.01 (s, 2 H, PhCH2) 6.78 (d, J = 9.2 Hz, 1 H, ArH), 7.26–7.42 (m, 6 H, 6 × ArH); 13C NMR (CDCl3; 100 MHz) δ 35.85, 51.92, 55.88, 74.89, 112.69, 112.70, 115.89, 127.55, 127.98, 128.09, 123.33, 128.35, 129.14, 137.28, 147.37, 152.09, 171.05 (CO); IR (neat) 2074 (w), 1736 (m, CO), 1635 (s), 1470 (m), 1273 (m), 1078 (m), 1004 (w), 747 (s) cm−1; MS (EI) m/z (relative intensity) 366 (M+ + 2, 65), 364 (M+, 65), 242 (4), 213 (20), 91 (100), 65 (10); HRMS (EI) calcd for C17H17BrO4: 364.0310, found 364.0307.
(±)-Methyl 2-[(2-benzyloxy-6-bromo-3-methoxyphenyl)]-3-hydroxy-3-(3,4-dimethoxy-phenyl)propanoate (17)
To a solution containing diisopropylamine (649.6 mg, 6.416 mmol, 1.1 equiv) in tetrahydrofuran (50 mL) n-butyllithium (397.9 mg, 6.416 mmol, 1.1 equiv) was added at 0 °C. After stirring for 30 min, the resultant mixture was cooled to −78 °C. Methyl phenyl acetate 8 (2.125 g, 5.837 mmol, 1.0 equiv) was then added to the reaction mixture, which was stirred at the same temperature for an additional 30 min. Finally, 3,4-dimethoxybenzaldehyde (4, 969.6 mg, 5.835 mmol, 1.0 equiv) was added to the reaction mixture and stirring was continued for 1 h. The reaction mixture was quenched with saturated aqueous NH4Cl (50 mL) and extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with a saturated aqueous NaCl solution (30 mL), dried over MgSO4(s), filtered and concentrated under reduced pressure to afford a residue. Purification of the residue by chromatography with a silica gel column (30% EtOAc in hexane as the eluent) gave (±)-17 (2.010 g, 3.792 mmol) as a white solid in a 65% yield: mp (recrystallized from EtOAc) 95–96 °C; TLC Rf 0.56 (30% EtOAc in hexane as the eluent); 1H NMR ((CD3)2CO; 400 MHz) δ 3.56 (s, 3 H, OCH3), 3.66 (s, 6 H, 2 × OCH3), 3.92 (s, 3 H, CO2CH3), 4.56 (br, 1 H), 4.72 (s, 2 H, PhCH2), 5.59 (d, 1 H, CHO), 6.20–6.69 (m, 3 H, 3 × ArH), 6.75 (s, 1 H, ArH), 6.86 (d, J = 8.4 Hz, 1 H, ArH), 7.15 (d, J = 8.4 Hz, 1 H, ArH), 7.37–7.44 (m, 3 H, 3 × ArH), 7.53–7.55 (d, J = 7.2 Hz, 1 H, ArH); 13C NMR ((CD3)2CO; 100 MHz) δ 52.45, 55.56, 55.78, 56.27, 56.28, 73.01, 74.31, 111.31, 111.42, 114.08, 119.88, 128.53, 128.54, 128.55, 128.58, 129.05, 129.06, 129.07, 132.02, 134.57, 134.58, 138.68, 149.18, 149.30, 151.95, 174.42 (CO); IR (neat) 3478 (br, OH), 2934 (s), 2829 (w), 1718 (s, CO), 1514 (m), 1465 (s), 1432 (m), 1027 (m), 805 (m) cm−1; MS (FAB) m/z 532 (M+ + 2, 9), 364 (20), 285 (35), 273 (25), 136 (30), 91 (100); HRMS (FAB) calcd for C26H27BrO7: 530.0940, found 530.0947.
(±)-Methyl trans-2-(3,4-dimethoxyphenyl)-4-bromo-7-methoxy-2,3-dihydrobenzo[b]-furan-3-carboxylate (18)
To a solution containing (±)-17 (915.6 mg, 1.727 mmol, 1.0 equiv) in dry dichloromethane (30 mL) iodotrimethylsilane (760.3 mg, 3.799 mmol, 2.2 equiv) was added via a syringe at 0 °C. After stirring at room temperature for 1 h, it was quenched with saturated aqueous NH4Cl (20 mL) and extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were washed with a saturated aqueous NaCl solution (15 mL), dried over MgSO4(s), filtered and concentrated under reduced pressure to afford a residue. Purification of the residue by chromatography with silica gel column (25% EtOAc in hexane as the eluent) gave (±)-18 (546.8 mg, 1.295 mmol) as a yellow gummy oil in a 75% yield: TLC Rf 0.64 (20% EtOAc in hexane as the eluent); 1H NMR (CDCl3; 400 MHz) δ 3.73 (s, 3 H, OCH3), 3.81 (s, 9 H, CO2CH3 + 2 × OCH3), 4.30 (d, J = 7.2 Hz, 1 H, ArCHCO2), 5.82 (d, J = 7.2 Hz, 1 H, ArOCHAr), 6.70 (d, J = 8.8 Hz, 1 H, ArH), 6.79 (d, J = 8.0 Hz, 1 H, ArH), 6.86–6.92 (m, 2 H, 2 × ArH), 6.95 (d, J = 8.8 Hz, 1 H, ArH); 13C NMR (CDCl3; 100 MHz) δ 52.74, 55.93, 55.95, 56.31, 58.09, 87.89, 108.90, 109.70, 111.18, 114.38, 118.29, 118.30, 124.49, 126.21, 131.78, 144.10, 149.29, 149.44, 171.43 (CO); IR (neat) 2069 (w), 1736 (m, CO), 1637 (s), 1517 (m), 1485 (m), 1263 (m), 1025 (s), 762 (w) cm−1; MS (EI) m/z (relative intensity) 424 (M+ + 2, 74), 422 (M+, 73), 392 (100), 390 (99), 343 (34), 311 (33), 284 (30), 84 (55); HRMS (EI) calcd for C19H19BrO6: 422.0365, found 422.0362.
(±)-tert-Butyl (2E)-3-[2-(3,4-Dimethoxyphenyl)-7-methoxy-3-methoxycarbonyl-2,3-dihydrobenzo[b]furan-4-yl]propenoate (19)
To a solution containing (±)-18 (151.2 mg, 0.3582 mmol, 1.0 equiv), Pd(OAc)2 (8.043 mg, 0.0358 mmol, 0.10 equiv) and PPh3 (28.17 mg, 0.1074 mmol, 0.30 equiv) in Et3N (362.1 mg, 3.582 mmol, 10.0 equiv) tert-butyl acrylate (229.5 mg, 1.791 mmol, 5.0 equiv) was added. After the reaction mixture was stirred under nitrogen at 110 °C for 16 h, it was quenched with saturated aqueous NH4Cl (20 mL) and extracted with Et2O (3 × 30 mL). The combined organic layers were washed with a saturated aqueous NaCl solution (20 mL), dried over MgSO4(s), filtered and concentrated under reduced pressure to afford a residue. Purification of the residue by chromatography with a silica gel column (20% EtOAc in hexane as the eluent) gave (±)-19 (136.4 mg, 0.2901 mmol) as a yellow foam in a 81% yield: mp (recrystallized from EtOAc) 101–102 °C; TLC Rf 0.50 (30% EtOAc in hexane as the eluent); 1H NMR (CDCl3; 400 MHz) δ 1.53 (s, 9 H, C(CH3)3), 3.84 (s, 3 H, OCH3), 3.89 (s, 6 H, 2 × OCH3), 3.97 (s, 3 H, CO2CH3), 4.52 (d, J = 5.8 Hz, 1 H, ArCHCO2), 6.05 (d, J = 5.8 Hz, 1 H, ArOCHAr), 6.27 (d, J = 16.0 Hz, 1 H, Ph–CCH–CO2), 6.86–6.96 (m, 4 H, 4 × ArH), 7.24 (d, J = 8.4 Hz, 1 H, ArH), 7.66 (d, J = 16.0 Hz, 1 H, Ph–CHC–CO2); 13C NMR (CDCl3; 100 MHz) δ 28.07, 28.08, 28.09, 52.70, 55.83, 55.84, 56.01, 56.02, 80.25, 87.44, 108.69, 111.04, 112.84, 117.98, 119.78, 120.24, 124.68, 124.75, 132.27, 139.59, 145.77, 148.26, 149.12, 149.18, 166.11 (CO), 171.84 (CO); IR (neat) 2972 (w), 2936 (w), 1702 (s, CO), 1627 (m, CO), 1589 (s), 1483 (s), 1276 (s), 1146 (s), 1084 (m), 971 (m), 805 (s) cm−1; MS (EI) m/z (relative intensity) 470 (M+, 64), 440 (8), 414 (52), 382 (32), 337 (100), 309 (23), 239 (16), 231 (23); HRMS (EI) calcd for C26H30O8: 470.1941, found 470.1944.
Methyl 4-hydroxymethyl-7-methoxy-2-(3,4-dimethoxyphenyl)benzo[b]furan-3-carboxylate (20)
To a solution containing aldehyde 11 (51.20 mg, 0.1383 mmol, 1.0 equiv) in dry methanol (10 mL) 10% Pd/C (14.72 mg, 0.1383 mmol, 1.0 equiv) was added. The flask was evacuated and back-filled with a H2 balloon three times and allowed to stir under a H2(g) atmosphere at room temperature for 3 h. Afterwards, the reaction mixture was filtered through Celite (rinsing several times with methanol) and concentrated under reduced pressure to afford a residue. Purification of the residue by chromatography with a silica gel column (40% EtOAc in hexane as the eluent) gave 20 (46.12 mg, 0.1239 mmol) as white solids in a 90% yield: mp (recrystallized from CH2Cl2) 98–99 °C; TLC Rf 0.37 (35% EtOAc in hexane as the eluent); 1H NMR (CDCl3; 400 MHz) δ 3.82 (s, 3 H, OCH3), 3.90 (s, 6 H, 2 × OCH3), 3.95 (s, 3 H, CO2CH3), 4.72 (s, 2 H, CH2O), 6.74 (d, J = 8.0 Hz, 1 H, ArH), 6.90 (d, J = 8.4 Hz, 1 H, ArH), 7.11 (d, J = 8.0 Hz, 1 H, ArH), 7.28–7.32 (m, 2 H, 2 × ArH); 13C NMR (CDCl3; 100 MHz) δ 52.18, 55.84, 55.85, 55.92, 63.43, 106.51, 108.90, 110.62, 111.50, 121.98, 122.15, 125.58, 126.18, 126.62, 143.50, 144.94, 148.50, 150.48, 158.70, 166.74 (CO); IR (neat) 3435 (br, OH), 2849 (w), 2056 (w), 1728 (s, CO), 1630 (s), 1507 (m), 1464 (m), 1265 (m), 1096 (w), 1052 (w), 738 (s) cm−1; MS (EI) m/z (relative intensity) 372 (M+, 77) 356 (100), 339 (43), 309 (31), 85 (29), 57 (66); HRMS (EI) calcd for C20H20O7: 372.1209, found 372.1208.
Methyl 4-methyl-7-methoxy-2-(3,4-dimethoxyphenyl)benzo[b]furan-3-carboxylate (21)
To a solution containing aldehyde 11 (65.28 mg, 0.1763 mmol, 1.0 equiv) in dry methanol (10 mL) 10% Pd/C (18.73 mg, 0.1760 mmol, 1.0 equiv) was added. The flask was evacuated and back-filled with a H2 balloon three times and allowed to stir under a H2(g) atmosphere at room temperature for 24 h. Afterwards, the reaction mixture was filtered through Celite (rinsing several times with methanol) and concentrated under reduced pressure to afford a residue. Purification of the residue by chromatography with a silica gel column (40% EtOAc in hexane as the eluent) gave 21 (53.36 mg, 0.1498 mmol) as a yellow foam in a 85% yield: mp (recrystallized from EtOAc) 88–89 °C; TLC Rf 0.46 (20% EtOAc in hexane as the eluent); 1H NMR (CDCl3; 400 MHz) δ 2.47 (s, 3 H, CH3), 3.88 (s, 3 H, OCH3), 3.91 (s, 3 H, OCH3), 3.93 (s, 3 H, OCH3), 3.97 (s, 3 H, CO2CH3), 6.72 (d, J = 8.0 Hz, 1 H, ArH), 6.90–6.94 (m, 2 H, 2 × ArH), 7.39–7.42 (m, 2 H, 2 × ArH); 13C NMR (CDCl3; 100 MHz) δ 18.94, 52.10, 55.89, 55.96, 56.08, 106.94, 109.57, 110.80, 110.86, 121.13, 122.25, 123.13, 125.25, 127.28, 142.97, 143.41, 148.72, 150.28, 155.91, 166.40 (CO); IR (neat) 2917 (m), 2848 (w), 1723 (s, CO), 1629 (s), 1509 (s), 1463 (s), 1364 (w), 1262 (s), 1229 (s), 1123 (m), 1043 (m), 800 (m), 736 (m) cm−1; MS (EI) m/z (relative intensity) 356 (M+, 100), 325 (13), 97 (11), 85 (38), 71 (55), 57 (93); HRMS (EI) calcd for C20H20O6: 356.1260, found 356.1261.
Acknowledgements
We thank the National Science Council (grant No. 99-2113-M-007-008-MY3) and Ministry of Education of R.O.C. (grant No. 100N2011E1) for financial support. Valuable suggestions and information provided by Professor Ru Chih Huang of The Johns Hopkins University is greatly appreciated.References
- G. Johnson, S. G. Sunderwirth, H. Gibian, A. W. Coulter and F. X. Gassner, Phytochemistry, 1963, 2, 145–150 CrossRef CAS.
- (a) C. J. Kelley, J. R. Mahajan, L. C. Brooks, L. A. Neubert, W. R. Breneman and M. Carmack, J. Org. Chem., 1975, 40, 1804–1815 CrossRef CAS; (b) C. J. Kelley, R. C. Harruff and M. Carmack, J. Org. Chem., 1976, 41, 449–455 CrossRef CAS.
- H. Wagner, D. Wittmann and W. Schafer, Tetrahedron Lett., 1975, 8, 547–550 CrossRef.
- I. S. Abd-Elazem, H. S. Chen, R. B. Bates and R. C. C. Huang, Antiviral Res., 2002, 55, 91–106 CrossRef CAS.
- R. W. Jiang, K. M. Lau, P. M. Hon, T. C. W. Mak, K. S. Woo and K. P. Fung, Curr. Med. Chem., 2005, 12, 237–246 CAS.
- H. Kohda, O. Takeda, S. Tanaka, K. Yamasaki, A. Yamashita, T. Kurokawa and S. Ishibashi, Chem. Pharm. Bull., 1989, 37(5), 1287–1290 CrossRef CAS.
- Y. L. Lin, Y. Y. Chang, Y. H. Kuo and M. S. Shiao, J. Nat. Prod., 2002, 65, 745–747 CrossRef CAS.
- R. M. Jacobson and R. A. Raths, J. Org. Chem., 1979, 44, 4013–4014 CrossRef CAS.
- S. J. O'Malley, K. L. Tan, A. Watzke, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2005, 127, 13496–13497 CrossRef CAS.
- D. H. Wang and J. Q. Yu, J. Am. Chem. Soc., 2011, 133, 5767–5769 CrossRef CAS.
- J. Fischer, G. P. Savage and M. J. Coster, Org. Lett., 2011, 13, 3376–3379 CrossRef CAS and references cited therein.
- V. Dalla, P. Cotelle and J. P. Catteau, Tetrahedron Lett., 1997, 38, 1577–1580 CrossRef CAS.
- S. S. Jew, H. A. Kim, S. Y. Bae, J. H. Kim and H. G. Park, Tetrahedron Lett., 2000, 41, 7925–7928 CrossRef CAS.
- M. Nakajima, K. Tomioka and K. Koga, Tetrahedron, 1993, 49, 10807–10816 CrossRef CAS.
- T. Eicher, M. Ott and A. Speicher, Synthesis, 1996, 755–762 CrossRef CAS and references cited therein.
- R. C. Larock, E. K. Yum, M. J. Doty and K. K. C. Sham, J. Org. Chem., 1995, 60, 3270–3271 CrossRef CAS.
- (a) J. R. Hwu, K. S. Chuang, S. S. Chuang and S. C. Tsay, Org. Lett., 2005, 7, 1545–1548 CrossRef CAS; (b) J. R. Hwu, Y. C. Hsu, T. Josephrajan and S. S. Tsay, J. Mater. Chem., 2009, 19, 3084–3090 RSC.
- K. M. Markovich, V. Tantishaiyakul, A. Hamada, D. D. Miller, K. J. Romstedt, G. Shams, Y. Shin, P. F. Fraundorfer, K. Doyle and D. R. Feller, J. Med. Chem., 1992, 35, 466–479 CrossRef CAS.
- L. Juhasz, L. Szilagyi, S. Antus, J. Visy, F. Zsila and M. Simonyi, Tetrahedron, 2002, 58, 4261–4265 CrossRef CAS.
- A. E. Lanzilotti, R. Littell, W. J. Fanshawe, T. C. McKenzie and F. M. Lovell, J. Org. Chem., 1979, 44, 4809–4814 CrossRef CAS.
- For representative reports and a recent review, see: (a) G. H. Lee, E. B. Choi, E. Lee and C. S. Pak, J. Org. Chem., 1994, 59, 1428–1443 CrossRef CAS; (b) I. K. Youn, G. H. Yon and C. S. Pak, Tetrahedron Lett., 1986, 27, 2409–2410 CrossRef CAS; (c) E. A. Boyle, F. R. Mangan, R. E. Markwell, S. A. Smith, M. J. Thomson, R. W. Ward and P. A. Wyman, J. Med. Chem., 1986, 29, 894–898 CrossRef CAS; (d) G. H. Lee, I. K. Youn, E. B. Choi, H. K. Lee, G. H. Yon, H. C. Yang and C. S. Pak, Curr. Org. Chem., 2004, 8, 1263–1287 CrossRef CAS.
- M. S. M. Yuen, F. Xue, T. C. W. Mak and H. N. C. Wong, Tetrahedron, 1998, 54, 12429–12444 CrossRef CAS.
- C. L. Kao and J. W. Chern, J. Org. Chem., 2002, 67, 6772–6787 CrossRef CAS.
- T. Nakanishi, M. Suzuki, A. Mashiba, K. Ishikawa and T. Yokotsuka, J. Org. Chem., 1998, 63, 4235–4239 CrossRef CAS.
- For representative works, see: (a) K. Ogura, Y. Ito and G. Tsuchihashi, Bull. Chem. Soc. Jpn., 1979, 52, 2013–2022 CrossRef CAS and references cited therein; (b) E. Pinard, M. Gaudry, F. Henot and A. Thellend, Tetrahedron Lett., 1998, 39, 2739–2742 CrossRef CAS.
- S. Pinheiro, M. B. Lima, C. B. S. S. Goncalves, S. F. Pedarza and F. M. C. de Farias, Tetrahedron Lett., 2000, 41, 4033–4034 CrossRef CAS.
- A. B. Leduc and M. A. Kerr, Eur. J. Org. Chem., 2007, 237–240 CrossRef CAS.
- M. G. H. Russell, R. Baker, R. G. Ball, S. R. Thomas, N. N. Tsou and J. L. Castro, J. Chem. Soc., Perkin Trans. 1, 2000, 893–899 RSC.
- (a) C. B. Ziegler and R. F. Heck, J. Org. Chem., 1978, 43, 2941–2946 CrossRef CAS; (b) Y. Wu, H. Zhang, Y. Zhao, J. Zhao, J. Chen and L. Li, Org. Lett., 2007, 9, 1199–1202 CrossRef CAS.
- K. M. Rupprecht, J. Boger, K. Hoogsteen, R. B. Nachbar and J. P. Springer, J. Org. Chem., 1991, 56, 6180–6188 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra as well as HPLC chromatograms for compounds 11, (±)-12, (±)-13, (±)-5, 16, 8, (±)-17, (±)-18, (±)-19, 20, and 21. See DOI: 10.1039/c2ob25575h |
| This journal is © The Royal Society of Chemistry 2012 |