 Open Access Article
Open Access ArticleChemoenzymatic formal synthesis of (−)- and (+)-epibatidine†
Derek R. Boyda, Narain D. Sharmaa, Magdalena Kaika, Peter B. A. McIntyrea, Paul J. Stevenson*a and Christopher C. R. Allenb
aSchool of Chemistry and Chemical Engineering, Queen's University, Belfast BT9 5AG, UK. E-mail: p.stevenson@qub.ac.uk; Fax: +44 (0)-28-9097-4687; Tel: +44 (0) 28 9097 4426
bSchool of Biological Sciences, Queen's University, Belfast BT9 5AG, UK
First published on 18th January 2012
Abstract
The cis-dihydrocatechol, derived from enzymatic cis-dihydroxylation of bromobenzene using the microorganism Pseudomonas putida UV4, was converted into (−)-epibatidine in eleven steps with complete stereocontrol. In addition, an unprecedented palladium-catalysed disproportionation reaction gave the (+)-enantiomer of an advanced key intermediate employed in a previous synthesis of epibatidine.
1 Introduction
To date, over eight hundred alkaloids have been isolated from the defensive skin secretions of frogs of the Dendrobates family.1 Of these compounds, undoubtedly, the most important is (−)-epibatidine (Fig. 1) a potent nicotinic receptor antagonist isolated from the Ecuadorian poison frog Epipedobates tricolor.2 As an analgesic, (−)-epibatidine is much more potent than morphine. Since its mode of action is through the nicotinic receptor, and not the opiate receptor, it has provided a stimulus in the search of new analgesics based around structural elements of the (−)-epibatidine structure.3 Due to the scarcity of material, available from the natural source, (−)-epibatidine could only be assigned a tentative structure which was confirmed later by its synthesis.4 Subsequently, keen interest was generated in the synthesis of epibatidine and within two years of its structure elucidation eleven racemic syntheses were reported.5,6 To date, over fifty syntheses and formal syntheses of epibatidine have appeared in the literature and the work has also been reviewed.7 All the very early routes relied on separation of enantiomers to get enantioenriched material. These samples were subsequently used to: (a) confirm the absolute configuration of (−)-epibatidine and (b) investigate its much awaited detailed biological evaluation.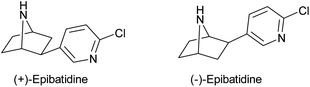 | ||
| Fig. 1 Structures of (+) and (−)-epibatidine. | ||
Nearly one quarter of the reported syntheses of epibatidine gave enantioenriched material without resolution. A wide variety of strategies have been employed to obtain optically active epibatidine including asymmetric desymmetrisation,8,9 asymmetric protonation,10 Diels–Alder reaction with chiral allenes,11 oxazolidinones12 and nitroso dienophiles,13,14 1,3-dipolar cycloaddition with sultam dienophiles,15 biocatalytic asymmetric monohydroxylation,16 1,4-addition to chiral enones derived from (−)-quinic acid,17 organocatalytic 1,4-addition to nitroalkenes,18 and addition of allyl zinc reagents to chiral imines.19 Although in most of these syntheses the stereoselectivity for installation of all three chiral centres was very high it could be further improved by recrystallisation, or recycling the unwanted stereoisomers, in none of the synthetic approaches was the innate stereoselectivity for generating all three chiral centres greater than 95% de/ee.
2 Results and discussion
Dioxygenase-catalysed oxidation of arene substrates provides a direct route to a wide range of enantiopure mono- and poly-hydroxylated bioproducts. Earlier studies of aromatic substrates, in these and other laboratories, using mutant strains (e.g. UV4, 39D) of the soil bacterium Pseudomonas putida and Escherichia coli recombinant strains, each containing toluene dioxygenase, have given access to an extensive range of over 400 metabolites.20 These enantiopure cis-dihydrodiols are ideal synthetic precursors for cyclohexane based natural products, as every carbon in the ring is fully functionalised and the biotransformations can be easily scaled up. Therefore it is not surprising that these new chiral pool derivatives are being extensively used in the synthesis of natural products,21 carbasugars,22 active pharmaceutical ingredients23 and as ligands in asymmetric catalysis.24 Starting from dihydrodiol 1 our chemoenzymatic route to (−)-epibatidine is outlined (Scheme 1). Chemoselective hydrogenation25,26 of the unsubstituted alkene bond gave the tetrahydrodiol 2. Previous X-ray studies have shown that in the crystalline state the hydroxyl group in cyclic allylic alcohols prefers to occupy a pseudo-axial position, to minimise allylic 1,2-strain with the substituent on the alkene.27 Analysis of the proton NMR spectrum of diol (−)-2 revealed proton H-1 to be a doublet of triplets (J 9.3, 3.9 Hz). The large diaxial coupling to H-6 also indicated that the C-1 hydroxyl group was equatorial and that the C-2 hydroxyl group was pseudo-axial in solution. As axial, or pseudo-axial alcohols occupy a more crowded position; they are much less reactive than equatorial alcohols in reactions that lead to an increase in steric bulk i.e. reactions involving the replacement of hydrogen attached to the oxygen with a larger group.28 Thus silylation, of diol (−)-2 was chemoselective with the homoallylic equatorial hydroxyl group forming the silyl ether 3. The silyl group in ether (−)-3 was found to be prone to scrambling, on storage or attempted purification, and as such was used immediately for the next step.![Reagents and conditions: (i) H2 (1.5 bar), Rh–G, MeOH, 2 h, rt, (90%); (ii) TBSCl, C6H5N, 12 h, rt (86%); (iii) trichloroacetyl isocyanate, CH2Cl2, 0.5 h, 0 °C then Na2CO3, Et2O : MeOH : H2O 4 : 4 : 1, 5 h, rt (90%); (iv) (a) Ph3P, Et3N, CBr4, CH2Cl2 −10 °C 1.5 h. (b) [3,3] sigmatropic rearrangement. (c) 1% MoCl2O2, CH2Cl2, t-BuOH, 16 h, rt (76% from 3; (v) 6-chloropyridin-3-ylboronic acid, 2% PdCl2(dppf), PhMe : EtOH : H2O 1 : 1 : 1, Na2CO3, 5 h, 100 °C (93%); (vi) TBAF, THF, 1 h (95%); (vii) PtO2, H2, 1 bar, EtOH, 5 h, rt (60%); (viii) Et3N, MsCl, CH2Cl2, 0 °C, 20 minutes (91%); (ix) LiBr, THF, 60 °C, 36 h (78%); (x) TFA, CH2Cl2 1.5 h, rt (94%); (xi) CHCl3, 55 °C, 72 h (95%).](/image/article/2012/OB/c2ob06904k/c2ob06904k-s1.gif) | ||
Scheme 1 Reagents and conditions: (i) H2 (1.5 bar), Rh–G, MeOH, 2 h, rt, (90%); (ii) TBSCl, C6H5N, 12 h, rt (86%); (iii) trichloroacetyl isocyanate, CH2Cl2, 0.5 h, 0 °C then Na2CO3, Et2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH:H2O 4:4:1, 5 h, rt (90%); (iv) (a) Ph3P, Et3N, CBr4, CH2Cl2 −10 °C 1.5 h. (b) [3,3] sigmatropic rearrangement. (c) 1% MoCl2O2, CH2Cl2, t-BuOH, 16 h, rt (76% from 3; (v) 6-chloropyridin-3-ylboronic acid, 2% PdCl2(dppf), PhMe:EtOH:H2O 1:1:1, Na2CO3, 5 h, 100 °C (93%); (vi) TBAF, THF, 1 h (95%); (vii) PtO2, H2, 1 bar, EtOH, 5 h, rt (60%); (viii) Et3N, MsCl, CH2Cl2, 0 °C, 20 minutes (91%); (ix) LiBr, THF, 60 °C, 36 h (78%); (x) TFA, CH2Cl2 1.5 h, rt (94%); (xi) CHCl3, 55 °C, 72 h (95%). :MeOH:H2O 4:4:1, 5 h, rt (90%); (iv) (a) Ph3P, Et3N, CBr4, CH2Cl2 −10 °C 1.5 h. (b) [3,3] sigmatropic rearrangement. (c) 1% MoCl2O2, CH2Cl2, t-BuOH, 16 h, rt (76% from 3; (v) 6-chloropyridin-3-ylboronic acid, 2% PdCl2(dppf), PhMe:EtOH:H2O 1:1:1, Na2CO3, 5 h, 100 °C (93%); (vi) TBAF, THF, 1 h (95%); (vii) PtO2, H2, 1 bar, EtOH, 5 h, rt (60%); (viii) Et3N, MsCl, CH2Cl2, 0 °C, 20 minutes (91%); (ix) LiBr, THF, 60 °C, 36 h (78%); (x) TFA, CH2Cl2 1.5 h, rt (94%); (xi) CHCl3, 55 °C, 72 h (95%). | ||
syn-Replacement of the alcohol oxygen by nitrogen with allylic rearrangement was efficiently achieved, with complete stereocontrol, using the Ichikawa allyl cyanate to isocyanate [3,3]-sigmatropic rearrangement.29 An attractive feature of this protocol is that it proceeds at low temperature and, in principle, the isocyanate intermediate 6 could be directly converted into any urethane N-protecting group by reaction with an appropriate alcohol. Employing Ichikawa's conditions, alcohol (−)-3 was converted to urethane 4 in high yield. Urethane 4 was then dehydrated to furnish intermediate cyanate 5 which, under the reaction conditions, underwent a rapid [3,3]-sigmatropic rearrangement to furnish the intermediate isocyanate 6 as the sole diastereoisomer. Our first choice of N-protecting group was Boc but, initially, the isocyanate 6 proved to be completely unreactive towards t-butanol or its metal alkoxides. However, using the newly developed Bruckner protocol,30 with molybdenum (VI) dichloride dioxide as a catalyst, the isocyanate 6 reacted smoothly with t-butanol to give the Boc derivative (+)-7. This approach allowed the conversion of urethane 4 to Boc-derivative 7 as a one pot operation, without isolation of the sensitive isocyanate 6, in an overall yield of 76%.
Suzuki cross coupling31 of alkenyl bromide (+)-7 with commercially available 6-chloropyridin-3-ylboronic acid proceeded smoothly and gave compound (+)-8. Fluoride-induced removal of the O-silyl protecting group gave allylic alcohol (+)-9. The next step involved catalytic stereoselective reduction of the hindered, trisubstituted alkene with retention of the chloropyridine group. It was envisaged that the N and O substituents would direct addition of hydrogen to the alkene face anti to these groups.32 In a previous synthesis of racemic epibatidine it was reported that the 6-chloropyridine was prone to hydrogenolysis32 but Kibayashi et al.13 and Baker et al.6 have successfully reduced an alkene bond in the presence of a 6-chloropyridyl group. In our hands when hydrogenation of alkene (+)-9, using platinum oxide as catalyst in ethanol as solvent, was allowed to proceed to full conversion of alkene (+)-9, only intractable material was produced. However, when the hydrogenation reaction was stopped after ca. 70% completion, the desired reduction product (−)-10 was obtained as a single diastereoisomer; its properties were identical to those previously reported by Evans.12 Subsequently, using the Evans protocol, compound (−)-10 was converted in a two step sequence to bromide (−)-12 with inversion of configuration. On removal of the N-protecting group, compound 13 cyclised to afford (−)-epibatidine, whose spectral and chiroptical properties were identical to those previously reported.12,13
Including the initial biotransformation, this synthesis, overall, employed five reactions which generated six new stereocentres to achieve the desired stereochemistry at the final three chiral centres in (−)-epibatidine. In each of these stereo defining reactions the product formed was a single stereoisomer, as determined by NMR spectroscopy, making the synthesis completely stereoselective.
A potential shortcoming of the chemoenzymatic approach is that it often leads to only one of the two possible enantiomers. As (−)-epibatidine has pseudo symmetry in its structure, the shift of the chloropyridine substituent to the adjacent methylene carbon will yield the opposite enantiomer i.e. (+)-epibatidine (Fig. 1). In practice this could be achieved by shifting the bromine substituent in intermediate (+)-7 to the adjacent alkene carbon (Scheme 1).
During our attempts to carry out Suzuki cross coupling of allylic alcohol (−)-14 with 6-chloropyridin-3-yl boronic acid, an unusual high yielding reaction was observed which resulted in the formation of ketone (−)-15 (Scheme 2). Formally, the alcohol was oxidised to a ketone and the carbon bromine bond was simultaneously reduced (disproportionation).
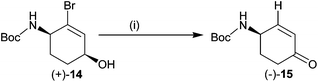 | ||
| Scheme 2 Reagents and conditions: (i) 10% PdCl2(dppf), PhMe:EtOH:H2O 1:1:1, Na2CO3, 6 h, 100 °C (82%). | ||
Further investigation revealed that ketone (−)-15 was formed even in the absence of 6-chloropyridin-3-yl boronic acid but the palladium catalyst and the base were both essential components of the reaction. It is presumed that the reaction involves a palladium-catalysed migration of the double bond to give the enol of the ketone which rapidly tautomerises to form a β-bromoketone. The ketone then loses hydrogen bromide under the basic reaction conditions to yield enone (−)-15 which is the enantiomer of the intermediate used by Trost in the first asymmetric synthesis of epibatidine. In the absence of a deuterium labelling experiment, the proposed disproportionation (migration/tautomerisation/dehydrobromination) mechanism for the formation of enone (−)-15 remains speculative. A bromine atom can be subsequently introduced, regioselectively, into the α-position of enone (−)-15 as described by Trost and Cook.8 This novel disproportionation reaction thus constitutes a formal chemoenzymatic synthesis of (+)-epibatidine.
In conclusion, we have demonstrated that bromobenzene cis-dihydrodiol (+)-1 can be converted, with complete stereocontrol of the three new chiral centres, to (−)-epibatidine. A newly discovered palladium-catalysed disproportionation reaction allows entry into the enantiomeric series. Since a wide range of aromatic boronic acids are commercially available this route is ideal for preparing epibatidine analogues for further biological evaluation.
3 Experimental section
cis-Dihydrodiol (+)-1, was obtained from the biotransformation of bromobenzene, using the dioxygenase enzymes present in the whole cell cultures of P. putida UV4 and the reduction to (−)-2 is as detailed elsewhere.33,26 All reagents were used as purchased from Aldrich and 6-chloropyridin-3-ylboronic acid was donated by Frontier Scientific. 1H-NMR and 13C-NMR spectra were recorded on Bruker Avance DPX-300 and AV-400 instruments and coupling constants are reported in Hz. Mass spectra were run at 70 eV, on an AE1-MS90 Mass Spectrometer updated by VG Autospec, using a heated inlet system. Accurate molecular weights were determined, by the peak matching method, with perfluorokerosene as standard. For optical rotation ([α]D) measurements (ca. 20 °C, 10−1 degree cm2 g−1) a Perkin-Elmer 214 polarimeter was used. Flash chromatography and preparative layer chromatography (PLC) were performed on Merck Kieselgel type 60 (250–400 mesh) and PF254/366 respectively. Merck Kieselgel type 60F254 analytical plates were used for TLC.(−)- (1S,6S)-2-Bromo-6-(tert-butyldimethylsilyloxy)cyclohex-2-enol 3
tert-Butyldimethylsilyl chloride (1.76 g, 0.014 mol) was added to a solution of cis-tetrahydrodiol (−)-2 (2.26 g, 0.012 mol) in dry pyridine (5 mL) under a nitrogen atmosphere. The reaction mixture was allowed to stir overnight at room temperature. Excess of pyridine was removed under reduced pressure, the residue dissolved in EtOAc (50 mL) and the solution washed, successively, with a sat. solution of NaHCO3 (15 mL), brine (15 mL) and dried (Na2SO4). Removal of solvent gave TBS derivative 3 as a light yellow oil (3.10 g, 86%), which was used for the next stage without further purification. Rf 0.41 (10% EtOAc in hexanes); [α]D −43.7 (c 1.1, CHCl3); HRMS (ES): found [M + H]+ 307.0732, C12H24O2BrSi requires 307.0729; 1H-NMR (400 MHz, CDCl3): δ 0.11 (3H, s, SiMe2), 0.12 (3H, s, SiMe2), 0.91 (9H, s, CMe3), 1.63 (1H, m, 5-H), 1.83 (1H, m, 5′-H), 2.04 (1H, m, 4-H), 2.22 (1H, m, 4′-H), 2.78 (1H, d, J 3.7 Hz, OH), 3.94 (1H, ddd, J 10.2, 4.1, 3.4, 6-H), 4.10 (1H, appar t, J 3.6, 1-H), 6.20 (1H, dd, J 4.8, 3.3, 3-H); 13C-NMR (CDCl3, 100 MHz): δ −4.8, −4.5, 18.1, 25.3, 25.5, 25.8 (3C), 70.8, 72.3, 121.9, 132.4; LRMS (EI): m/z 277 (30), 215 (25), 149 (100), 104 (65).(+)-tert-Butyl-(1R,4S)-2-bromo-4-(tert-butyldimethylsilyloxy)cyclohex-2-enylcarbamate 7
Trichloroacetyl isocyanate (0.88 mL, 7.43 mmol) was added to an ice cooled solution of silyl ether (−)-3 (1.18, 3.84 mmol) in CH2Cl2 (25 mL). After stirring the mixture for 0.5 h at 0 °C the solvent was evaporated, the residue dissolved in a mixture of MeOH (20 mL) and Et2O (5 mL) and the solution stirred at room temperature for 5 h with aq. K2CO3 solution (1 M, 20 mL). Most of the methanol was removed under reduced pressure and the aqueous reaction mixture extracted with Et2O (3 × 30 mL). The combined organic extract was washed with brine (60 mL), dried (MgSO4) and concentrated under reduced pressure to give urethane 4 as an off-white solid (1.25 g, 90%), which was used in the next step without further purification.To a solution of urethane 4 (1.25 g, 3.45 mmol) in anhydrous CH2Cl2 (20 mL) were added triphenylphoshine (2.71 g, 10.3 mmol) and Et3N (2.79 mL, 20.01 mmol). The mixture was cooled to −10 °C and CBr4 (3.43 g, 10.4 mmol) solution in dry CH2Cl2 (30 mL) was added to it drop-wise over 1 h. The reaction mixture was kept stirring at −10 °C for a further 1.5 h, freshly distilled tert-butanol (0.51 g, 6.90 mmol) and molybdenum (VI) dichloride dioxide (6.9 mg, 0.034 mmol) were then added sequentially and it was allowed to warm to room temperature. After stirring the mixture for another 16 h, it was washed with 10% aq. NaHCO3 (20 mL), saturated brine (20 mL), dried (Na2SO4) and concentrated in vacuo. The resulting residue was purified by flash chromatography (5% EtOAc in hexanes) to yield compound (+)-7 as a clear oil (1.19 g, 76%, 2 steps). Rf 0.45 (10% EtOAc in hexanes); [α]D +18.3 (c 0.7, CHCl3); HRMS (ES): found [M + H]+ 406.1413, C17H33BrNO3Si requires 406.1408; 1H-NMR (400 MHz, CDCl3): δ 0.07 (3H, s, SiMe2), 0.08 (3H, s, SiMe2), 0.89 (9H, s, SiCMe3), 1.46 (9H, s, OCMe3), 1.49–1.63 (1H, m, 5-H), 1.79–1.97 (3H, m, 5′-H, 6-H, 6′-H), 4.17 (1H, dddd, J 8.9, 5.0, 3.1, 1.4, 4-H), 4.28 (1H, m, 1-H), 4.78 (1H, d, J 8.7, NHBoc), 6.13 (1H, ddd, J 3.0, 0.9, 0.9, 3-H); 13C-NMR (CDCl3, 100 MHz): δ −4.6, −4.6, 18.2, 25.9 (3C), 28.0, 28.1, 28.5 (3C), 51.7, 68.4, 79.9, 125.3, 137.3, 155.4; LRMS (EI): m/z 292 (15), 248 (10), 118 (25), 74 (100).
(+)-tert-Butyl-(1R,4S)-4-(tert-butyldimethylsilyloxy)-2-(6-chloropyridin-3-yl)cyclohex-2-enylcarbamate 8
A mixture of 6-chloropyridin-3-ylboronic acid (0.46 g, 2.95 mmol), EtOH (10 mL) and aq. Na2CO3 (2M, 8 mL) was added to a round bottom flask containing a solution of tert-butyl (1R,4S)-carbamate (+)-7 (1.00 g, 2.46 mmol) in toluene (10 mL). After flushing the contents of the flask with a stream of nitrogen, PdCl2(dppf) (40 mg, 0.055 mmol) was added and the mixture was refluxed with stirring for 5 h under nitrogen. The reaction mixture was transferred into a separating funnel, the organic layer was separated and the aq. layer extracted with EtOAc (3 × 20 mL). The combined organic extract was washed with brine (50 mL), dried (Na2SO4) and concentrated in vacuo to give a dark brown oil. Purification of the oil by flash chromatography (5–10% EtOAc in hexanes) gave compound (+)-8 as a white crystalline solid (1.00 g, 93%); m.p. 106–107 °C; [α]D +36.6 (c 1.2, CHCl3); Rf 0.3 (10% EtOAc in hexanes); HRMS (ES): found [M + H]+ 439.2192, C22H36ClN2O3Si requires 439.2184; 1H-NMR (400 MHz, CDCl3): δ 0.12 (3H, s, SiMe2), 0.13 (3H, s, SiMe2), 0.93 (9H, s, SiCMe3), 1.37 (9H, s, OCMe3), 1.56–1.68 (1H, m, 5-H), 1.79–2.02 (3H, m, 5′-H, 6-H, 6′-H), 4.32 (1H, ddd, J 8.4, 5.6, 2.8, 4-H), 4.67 (2H, m, 1-H, NHBoc), 6.11 (1H, d, J 2.7, 3-H), 7.27 (1H, d, J 8.5, Ar-5-H), 7.69 (1H, dd, J 8.5, 2.6, Ar-4-H), 8.44 (1H, d, J 2.6, Ar-2-H); 13C-NMR (100 MHz, CDCl3): δ −4.6, −4.4, 18.3, 26.0 (3C), 27.5, 28.0, 28.4 (3C), 45.3, 67.3, 79.9, 123.8, 133.8, 134.7, 134.7, 136.4, 147.7, 150.5, 155.2; LRMS (ES): m/z 877 (30), 439 (100), 383 (10).(+)-tert-Butyl-(1R,4S)-2-(6-chloropyridin-3-yl)-4-hydroxycyclohex-2-enylcarbamate 9
To an ice cooled solution of carbamate (+)-8 (0.90 g, 2.05 mmol) in THF (8 mL) was added a THF solution of tetrabutylammonium fluoride (1 M, 4.48 mL, 4.51 mmol). The reaction mixture was stirred at room temperature for 1 h under nitrogen. Saturated aq. NH4Cl (15 mL) was then added and the mixture extracted with EtOAc (3 × 15 mL). The combined organic extract was dried (Na2SO4), concentrated in vacuo and the crude product purified by flash chromatography (20% EtOAc in hexanes, then 50% EtOAc in hexanes) to give compound (+)-9 as an off-white low melting solid (0.63 g, 95%); m.p. 54–56 °C; [α]D +80.3 (c 0.6, CHCl3); Rf 0.22 (50% EtOAc in hexanes); HRMS (ES): found [M + H]+ 325.1308, C16H22ClN2O3 requires 325.1319; 1H-NMR (400 MHz, CDCl3): δ 1.37 (9H, s, OCMe3), 1.63–1.71 (2H, m, 5-H, OH) 1.84–1.95 (1H, m, 6-H), 1.96–2.10 (2H, m, 5′-H, 6′-H), 4.37 (1H, m, 4-H), 4.63–4.70 (2H, m, 1-H, NHBoc), 6.23 (1H, d, J 3.0, 3-H), 7.28 (1H, d, J 8.4, Ar-5-H), 7.70 (1H, dd, J 8.4, 2.5, Ar-4-H), 8.44 (1H, d, J 2.5, Ar-2-H); 13C-NMR (100 MHz, CDCl3): δ 27.2, 28.0, 28.3 (3C), 45.6, 66.3, 80.0, 123.8, 133.0, 133.2, 136.2, 136.4, 147.6, 150.6, 155.2; LRMS (ES): m/z 649 (10), 325 (100), 269 (40), 217 (25).(−)-tert-Butyl-(1R,2R,4S)-2-(6-chloropyridin-3-yl)-4-hydroxycyclohexylcarbamate 10
To a solution of allylic alcohol (−)-9 (100 mg, 0.304 mmol), in EtOH (7 mL) was added PtO2 (10 mg, 10% by wt) and the mixture stirred for 5 h under 1 atm. of hydrogen at room temperature. When the reaction had progressed to 70% completion, as monitored by 1H-NMR spectroscopy, the catalyst was removed by filtration and the filtrate concentrated in vacuo. The residue obtained was purified by flash chromatography (60% EtOAc in hexanes) to give compound (−)-10, a white solid (60 mg, 60%) and unreacted starting material (24 mg, 24%); [α]D −41.5 (c 0.5, CHCl3); Rf 0.15 (50% EtOAc in hexanes); HRMS (ES): found [M + Na]+ 349.1290, C16H23ClN2O3Na requires 349.1295; 1H-NMR (400 MHz, CDCl3): δ 1.26 (9H, s, OCMe3), 1.37–1.39 (1H, m, 6-H), 1.65–1.81 (2H, m, 3-H, 5-H), 1.93–2.12 (3H, m, 3′-H, 5′-H, 6′-H), 2.91 (1H, d, J 13.5, 2-H), 3.79 (1H, m, 4-H), 4.03 (1H, br s, 1-H), 4.72 (1H, d, J 9.6, NHBoc), 7.24 (1H, d, J 8.2, Ar-5-H), 7.50 (1H, dd, J 8.2, 2.4, Ar-4-H), 8.22 (1H, d, J 2.0, Ar-2-H); 13C-NMR (100 MHz, CDCl3): δ 28.2 (3C), 29.6, 29.8, 34.4, 42.2, 48.5, 69.8, 79.6, 123.9, 136.2, 138.4, 148.7, 149.8, 155.0; LRMS (ES): m/z 1387 (3), 1061 (13), 739 (10), 675 (28), 556 (70), 413 (30), 349 (100), 293 (40), 239 (25). The spectral characteristics of compound (−)-10 were identical to those reported. 12,13(−)-(1S,3R,4R)-4-(tert-Butoxycarbonylamino)-3-(6-chloropyridin-3-yl)cyclohexyl methanesulphonate 11
An ice cooled solution of alcohol (−)-10 (50 mg, 0.15 mmol) in dry CH2Cl2 (4 mL) containing Et3N (0.31 mL, 2.20 mmol) was treated with methanesulphonyl chloride (0.15 mL, 1.97 mmol). After stirring the reaction mixture for 20 minutes at 0 °C, it was diluted with EtOAc (20 mL) and then washed successively with sat. NaHCO3 (2 × 10 mL), sat. NH4Cl (2 × 10 mL) and brine (20 mL). The organic layer was dried (Na2SO4), concentrated in vacuo, and the yellow oil obtained was purified by PLC (50% EtOAc in hexanes) to furnish mesylate (−)-11 as a colourless oil (56 mg, 91%); [α]D −56.7 (c 0.96, CH2Cl2); Rf 0.2 (50% EtOAc in hexanes); HRMS (ES): found [M+H]+ 405.1257, C17H26ClN2O5S requires 405.1251; 1H-NMR (400 MHz, CDCl3): δ 1.25 (9H, s, OCMe3), 1.65–1.69 (1H, m, 6-H), 1.75–1.86 (1H, m, 5-H), 1.96–2.09 (2H, m, 2-H, 5′-H), 2.15–2.23 (1H, m, 6′-H), 2.24–2.34 (1H, m, 2′-H), 2.97 (1H, br d, J 13.8, 3-H), 3.03 (3H, s, SO2Me), 4.06 (1H, br s, 4-H), 4.76 (1H, dddd, J 11.2, 11.2, 5.3, 5.3, 1-H), 7.26 (1H, m, Ar-5-H), 7.50 (1H, dd, J 8.3, 2.5, Ar-4-H), 8.22 (1H, br s, Ar-2-H); 13C-NMR (100 MHz, CDCl3): δ 14.3, 21.1, 22.7, 28.2 (3C), 31.8, 39.0, 42.2, 60.5, 79.2, 124.0, 135.1, 138.2, 148.6, 150.1, 171.3; LRMS (ES): m/z 405 (98), 239 (32), 217 (100). The spectral characteristics of compound (−)-11 were identical to those reported.12,13(−)-tert-Butyl (1R,2R,4R)-4-bromo-2-(6-chloropyridin-3-yl)cyclohexylcarbamate 12
Anhydrous LiBr (129 mg, 1.48 mmol) was added to a solution of mesylate (−)-11 (30 mg, 0.07 mmol) in dry THF (5 mL) and the mixture heated at 60 °C for 36 h. The reaction mixture was cooled, diluted with EtOAc (40 mL), washed with brine (2 × 30 mL), dried (Na2SO4) and concentrated to give a yellow oil. Purification of the oil by PLC (20% EtOAc in hexanes) yielded compound (−)-12 as a white crystalline solid (23 mg, 78%); [α]D −26.2 (c 1, CHCl3); Rf 0.36 (20% EtOAc in hexanes); HRMS (ES): found [M + Na]+ 411.0430, C16H22ClN2O2NaBr requires 411.0451; 1H NMR (CDCl3): δ 1.26 (9H, br s, OCMe3), 1.84 (1H, m, 6-H), 1.96 (1H, m, 5-H), 2.06 (1H, m, 5′-H), 2.13–2.36 (3H, m, 6′-H, 3-H, 3′-H), 3.52 (1H, dt, J 12.2, 3.4, 2-H), 4.10 (1H, br s, 1-H), 4.62 (1H, br s, NHBoc), 4.81 (1H, m, 4-H), 7.27 (1H, d, J 2.3, Ar-5-H), 7.52 (1H, dd, J 8.5, 2.3, Ar-4-H), 8.22 (1H, br s, Ar-2-H); 13C-NMR (CDCl3, 100 MHz): δ 26.5, 28.1 (3C), 29.1, 33.3, 38.4, 49.3, 51.6, 79.9, 123.9, 136.1, 138.6, 148.7, 149.9, 155.0; LRMS (ES): m/z 507 (20), 467 (30), 413 (100), 239 (60). The spectral characteristics of compound (−)-12 were identical to those reported.12,13(1R,2R,4R)-4-Bromo-2-(6-chloropyridin-3-yl)cyclohexanamine 13
Trifluoroacetic acid (0.4 mL) was added to a solution of protected amine (−)-12 (23 mg, 0.06 mmol), in dry CH2Cl2 (3 mL) and the mixture stirred at room temperature for 1.5 h. The reaction mixture was diluted with CHCl3 (25 mL), washed with 10% aq. K2CO3 (25 mL) and the organic layer separated. The aq. layer was extracted with CHCl3 (15 mL), the combined organic extract dried (K2CO3) and concentrated under reduced pressure to give the crude amine bromide 13 as light yellow oil (16 mg, 94%). It showed spectral characteristics identical to those reported12,13 and was used for the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 1.67 (1H, dq, J 13.5, 3.4, 6-H), 1.91 (1H, m, 5-H), 1.98 (1H, m, 3-H), 2.26 (1H, tt, J 13.4, 3.1, 6′-H), 2.35 (1H, tt, J 13.5, 3.3, 5′-H), 2.67 (1H, ddd, J 14.1, 12.8, 3.1, 3-H), 3.35 (1H, m, 1-H), 3.44 (1H, dt, J 12.8, 3.1, 2-H), 4.87 (1H, t, J 3.1, 4-H), 7.29 (1H, d, J 8.2, Ar-5-H), 7.53 (1H, dd, J 8.2, 2.6, Ar-4-H), 8.28 (1H, d, J 2.6, Ar-2-H); 13C-NMR (CDCl3,100 MHz): δ 27.8, 28.6, 29.8, 32.0, 50.1, 54.3, 124.2, 137.5, 138.4, 149.5, 149.9.(−)-Epibatidine
A solution of amine bromide 13 (16 mg, 0.055 mmol) in CHCl3 (3 mL) was heated in a sealed tube at 55 °C for 3 days. The reaction mixture was diluted with CHCl3 (30 mL) washed with 10% aqueous K2CO3 (15 mL) and the organic layer separated. The aq. layer was extracted with CHCl3 (20 mL), the combined organic extract dried (K2CO3) and concentrated to give a light yellow oil. Purification by PLC (95:5:0.5, CH2Cl2:MeOH:NH4OH) furnished (−)-epibatidine as a white solid (11 mg, 95%); [α]D −6.1 (c 1.0, CHCl3); HRMS (LCMS): found [M + H]+ 209.0840, C11H14ClN2 requires 209.0840; 1H-NMR (400 MHz, CDCl3): δ 1.48–1.66 (5H, m, 3-H, 5-H, 5′-H, 6-H, 6′-H), 1.91 (1H, dd, J 12.2, 9.3, 3′-H), 2.27 (1H, br s, NH), 2.77 (1H, dd, J 9.3, 4.9, 2-H), 3.57 (1H, br d, J 1.9, 1-H), 3.80 (1H, t, J 4.3, 4-H), 7.23 (1H, d, J 8.3, Ar-5-H), 7.76 (1H, dd, J 8.3, 2.5, Ar-4-H), 8.27 (1H, d, J 2.5, Ar-2-H); 13C-NMR (100 MHz, CDCl3): δ 30.2, 31.4, 40.4, 44.7, 56.6, 62.9, 124.1, 137.8, 141.1, 148.9, 149.1. LRMS (ES): m/z 209 (62), 192 (100), 166 (54), 126 (82). The spectral data of (−)-epibatidine was in good agreement with literature values.12,13(+)-tert-Butyl (1R,4S)-2-bromo-4-hydroxycyclohex-2-enylcarbamate 14
To an ice cooled solution of silylether (+)-7 (102 mg, 0.192 mmol) in THF–H2O (2 mL, 3:1) was added a THF solution of tetrabutylammonium fluoride (1 M, 1 mL, 1.0 mmol) and the mixture stirred overnight at room temperature. The solvents were removed under reduced pressure and the residual light yellow oil purified by PLC (40% EtOAc in hexanes) to give compound (+)-14 as a clear oil (46 mg, 82%); [α]D +36.6 (c 0.50, CHCl3); Rf 0.31 (40% EtOAc in hexanes); HRMS (ES): found [M + Na]+ 314.0352, C11H18NO3BrNa requires 314.0368; 1H-NMR (400 MHz, CDCl3): δ 1.47 (9H, s, CMe3), 1.62 (1H, m, 5-H), 1.97 (3H, m, 5′-H, 6-H, 6′-H), 4.22 (2H, m, 1-H, 4-H), 4.86 (1H, s, NHBoc), 6.29 (1H, d, J 2.69, 3-H); 13C-NMR (100 MHz): δ 27.5, 28.0, 28.5 (3C), 52.0, 67.4, 80.1, 127.3, 135.9, 155.4; LRMS (EI): m/z 279 (5) 277 (5), 234 (10), 236 (10), 217 (30), 175 (10), 156 (100), 94 (50).(−)- (R)-tert-Butyl 4-oxocyclohex-2-enylcarbamate 15
A slow stream of argon gas was bubbled through a mixture of allylic alcohol (+)-14 (32 mg, 0.11 mmol), toluene (1 mL) and aq. Na2CO3 (2 M, 0.4 mL) to displace any oxygen present in the solution. PdCl2(dppf) (9.4 mg, 0.013 mmol) was added to the mixture under a flow of argon. The reaction mixture was then heated under reflux with stirring for 6 h. The solvent was removed under reduced pressure, the residue taken up in EtOAc (5 mL), washed with brine (2 × 5 mL), dried (Na2SO4) and concentrated to give the crude product 15 as a yellow oil. Purification by PLC (20% EtOAc in hexane) yielded the enone (−)-15 as a white solid (19 mg, 82%); m.p. 117–118 °C, lit. m.p. 117–118; Rf 0.21 (20% EtOAc in hexane); [α]D +120.0, (c 0.50, CH2Cl2); lit. (ent) [α]D −123.6 (c 1.14, CH2Cl2);8 HRMS (EI): found (M+−tBu) 155.0587, C11H17NO3 requires 155.0577; 1H-NMR (400 MHz, CDCl3): δ 1.48 (9H, s, CMe3), 1.89 (1H, m, 6-H), 2.33 (1H, m, 6′-H), 2.50 (2H, m, 5-H, 5′-H), 4.53 (1H, br m, 1-H), 4.66 (1H, br m, NHBoc), 6.01 (1H, ddd, J 10.2, 2.3, 0.8, 3-H), 6.83 (1H, ddd, J 10.2, 2.4, 1.6, 2-H); 13C-NMR (100 MHz): δ 28.7 (3C), 30.4, 36.3, 47.4, 66.0, 130.7, 151.2, 155.4, 198.4. LRMS (EI): m/z 155 (48), 138 (20), 95 (50), 94 (100), 83 (48).Acknowledgements
We gratefully acknowledge financial support from DEL (PABMI), The Queen's University of Belfast and Dr Robert Cundell, Frontier Scientific, for a generous gift of 6-chloropyridin-3-ylboronic acid. We would also like to thank Dr Colin Mc Roberts and Mr Stewart Floyd for providing mass spectra.Notes and references
- J. W. Daly, T. F. Spande and H. M. Garraffo, J. Nat. Prod., 2005, 68, 1556 CrossRef CAS.
- T. F. Spande, H. M. Garraffo, M. W. Edwards, H. J. C. Yeh, L. Pannell and J. W. Daly, J. Am. Chem. Soc., 1992, 114, 3475 CrossRef CAS; H. M. Garraffo, T. F. Spande and M. Williams, Heterocycles, 2009, 79, 207 Search PubMed.
- B. Badio and J. W. Daly, Mol. Pharmacol., 1994, 45, 563 Search PubMed; J. W. Daly, H. M. Garraffo, T. F. Spande, M. W. Decker, J. P. Sullivan and M. Williams, Nat. Prod. Rep., 2000, 17, 131 RSC; P. Yogeeswari, D. Sriram, T. R. Bal and R. Thirumurugan, Nat. Prod. Res., 2006, 20, 497 Search PubMed.
- C. A. Broka, Tetrahedron Lett., 1993, 34, 3251 CrossRef CAS; E. J. Corey, T. P. Loh, S. Achyutharao, D. C. Daley and S. Sarshar, J. Org. Chem., 1993, 58, 5600 CrossRef CAS; S. R. Fletcher, R. Baker, M. S. Chambers, S. C. Hobbs and P. J. Mitchell, J. Chem. Soc., Chem. Commun., 1993, 1216 RSC; D. F. Huang and T. Y. Shen, Tetrahedron Lett., 1993, 34, 4477 CrossRef CAS; S. C. Clayton and A. C. Regan, Tetrahedron Lett., 1993, 34, 7493 CrossRef CAS.
- E. Albertini, A. Barco, S. Benetti, C. Derisi, G. P. Pollini, R. Romagnoli and V. Zanirato, Tetrahedron Lett., 1994, 35, 9297 CrossRef CAS; G. Pandey, T. D. Bagul and G. Lakshmaiah, Tetrahedron Lett., 1994, 35, 7439 CrossRef CAS; S. Y. Ko, J. Lerpiniere, I. D. Linney and R. Wrigglesworth, J. Chem. Soc., Chem. Commun., 1994, 1775 RSC; K. Sestanj, E. Melenski and I. Jirkovsky, Tetrahedron Lett., 1994, 35, 5417 CrossRef CAS; K. Okabe and M. Natsume, Chem. Pharm. Bull., 1994, 42, 1432 CAS; C. Szantay, Z. Kardosbalogh, I. Moldvai, E. Temesvarimajor and G. Blasko, Tetrahedron Lett., 1994, 35, 3171 CrossRef CAS.
- S. R. Fletcher, R. Baker, M. S. Chambers, R. H. Herbert, S. C. Hobbs, S. R. Thomas, H. M. Verrier, A. P. Watt and R. G. Ball, J. Org. Chem., 1994, 59, 1771 CrossRef CAS.
- H. F. Olivo and M. S. Hemenway, Org. Prep. Proced. Int., 2002, 34, 1 Search PubMed.
- B. M. Trost and G. R. Cook, Tetrahedron Lett., 1996, 37, 7485 CrossRef CAS.
- C. D. Jones, N. S. Simpkins and G. M. P. Giblin, Tetrahedron Lett., 1998, 39, 1023 CrossRef CAS; C. Pandey, S. K. Tiwari, R. S. Singh and R. S. Mali, Tetrahedron Lett., 2001, 42, 3947 Search PubMed; V. K. Aggarwal and B. Olofsson, Angew. Chem., Int. Ed., 2005, 44, 5516 CrossRef CAS.
- H. Kosugi, M. Abe, R. Hatsuda, H. Uda and M. Kato, Chem. Commun., 1997, 1857 RSC.
- H. Kimura, T. Fujiwara, T. Katoh, K. Nishide, T. Kajimoto and M. Node, Chem. Pharm. Bull., 2006, 54, 399 Search PubMed; M. Node, K. Nishide, T. Fujiwara and S. Ichihashi, Chem. Commun., 1998, 2363 RSC.
- D. A. Evans, K. A. Scheidt and C. W. Downey, Org. Lett., 2001, 3, 3009 CrossRef CAS.
- S. Aoyagi, R. Tanaka, M. Naruse and C. Kibayashi, J. Org. Chem., 1998, 63, 8397 CrossRef CAS.
- A. Hall, P. D. Bailey, D. C. Rees, G. M. Rosair and R. H. Wightman, J. Chem. Soc., Perkin Trans. 1, 2000, 329 RSC.
- I. Cabanal-Duvillard, J. F. Berrien, L. Ghosez, H. P. Husson and J. Royer, Tetrahedron, 2000, 56, 3763 CrossRef CAS; G. Pandey, J. K. Laha and G. Lakshmaiah, Tetrahedron, 2002, 58, 3525 CrossRef CAS.
- H. F. Olivo and M. S. Hemenway, J. Org. Chem., 1999, 64, 8968 CrossRef CAS.
- M. T. Barros, C. D. Maycock and M. R. Ventura, J. Chem. Soc., Perkin Trans. 1, 2001, 166 RSC.
- Y. Hoashi, T. Yabuta and Y. Takemoto, Tetrahedron Lett., 2004, 45, 9185 CrossRef CAS.
- C. L. K. Lee and T. P. Loh, Org. Lett., 2005, 7, 2965 CrossRef CAS.
- S. M. Resnick, K. Lee and D. T. Gibson, J. Ind. Microbiol., 1996, 17, 438 CrossRef CAS; T. Hudlicky, D. Gonzalez and D. T. Gibson, Aldrichimica Acta, 1999, 32, 35 CAS; D. R. Boyd and G. N. Sheldrake, Nat. Prod. Rep., 1998, 15, 309 RSC; D. T. Gibson and R. E. Parales, Curr. Opin. Biotechnol., 2000, 11, 236 CrossRef CAS; D. R. Boyd, N. D. Sharma and C. C. R. Allen, Curr. Opin. Biotechnol., 2001, 12, 564 CrossRef CAS; R. A. Johnson, Org. React., 2004, 63, 117 CAS; D. R. Boyd and T. D. H. Bugg, Org. Biomol. Chem., 2006, 4, 181 RSC; T. Hudlicky and J. W. Reed, Chem. Soc. Rev., 2009, 38, 3117 RSC; T. Hudlicky and J. W. Reed, Synlett, 2009, 685 CrossRef CAS.
- L. V. White, B. D. Schwartz, M. G. Banwell and A. C. Willis, J. Org. Chem., 2011, 76, 6250 Search PubMed; B. D. Schwartz, M. G. Banwell and I. A. Cade, Tetrahedron Lett., 2011, 52, 4526 Search PubMed; X. H. Ma, J. C. Jury and M. G. Banwell, Tetrahedron Lett., 2011, 52, 2192 Search PubMed; D. Bon, M. G. Banwell and A. C. Willis, Tetrahedron, 2011, 67, 5841 Search PubMed; M. G. Banwell, A. L. Lehmann, R. S. Menon and A. C. Willis, Pure Appl. Chem., 2011, 83, 411 Search PubMed; J. Duchek, D. R. Adams and T. Hudlicky, Chem. Rev., 2011, 111, 4223 Search PubMed; J. Duchek, T. G. Piercy, J. Gilmet and T. Hudlicky, Can. J. Chem., 2011, 89, 709 Search PubMed; S. Vshyvenko, J. Scattolon, T. Hudlicky, A. E. Romero and A. Kornienko, Bioorg. Med. Chem. Lett., 2011, 21, 4750 Search PubMed; D. R. Adams, C. Aichinger, J. Collins, U. Rinner and T. Hudlicky, Synlett, 2011, 1188 Search PubMed; D. R. Adams, C. Aichinger, U. Rinner and T. Hudlicky, Synlett, 2011, 725 CAS; T. Hudlicky, Pure Appl. Chem., 2010, 82, 1785 CrossRef CAS; T. K. Macklin and G. C. Micalizio, J. Am. Chem. Soc., 2009, 131, 1392 CrossRef CAS; D. R. Boyd, N. D. Sharma, C. A. Acaru, J. F. Malone, C. R. O'Dowd, C. C. R. Allen and P. J. Stevenson, Org. Lett., 2010, 12, 2206 CrossRef CAS.
- D. R. Boyd, N. D. Sharma, N. I. Bowers, G. B. Coen, J. F. Malone, C. R. O'Dowd, P. J. Stevenson and C. C. R. Allen, Org. Biomol. Chem., 2010, 8, 1415 RSC; D. R. Boyd, N. D. Sharma, N. M. Llamas, J. F. Malone, C. R. O'Dowd and C. C. R. Allen, Org. Biomol. Chem., 2005, 3, 1953 RSC.
- L. Werner, A. Machara and T. Hudlicky, Adv. Synth. Catal., 2010, 352, 195 CrossRef CAS; M. Matveenko, A. C. Willis and M. G. Banwell, Tetrahedron Lett., 2008, 49, 7018 CrossRef CAS.
- D. R. Boyd, N. D. Sharma, L. Sbircea, D. Murphy, J. F. Malone, S. L. James, C. C. R. Allen and J. T. G. Hamilton, Org. Biomol. Chem., 2010, 8, 1081 RSC; L. Sbircea, N. D. Sharma, W. Clegg, R. W. Harrington, P. N. Horton, M. B. Hursthouse, D. C. Apperley, D. R. Boyd and S. L. James, Chem. Commun., 2008, 5538 RSC; D. R. Boyd, N. D. Sharma, L. Sbircea, D. Murphy, T. Belhocine, J. F. Malone, S. L. James, C. C. R. Allen and J. T. G. Hamilton, Chem. Commun., 2008, 5535 RSC.
- D. R. Boyd, N. D. Sharma, M. Kaik, M. Bell, P. B. A. McIntyre, B. Kelly, C. Hardacre, P. J. Stevenson and C. C. R. Allen, Adv. Synth. Catal., 2011, 353, 2455 CrossRef CAS.
- D. R. Boyd, N. D. Sharma, N. M. Llamas, G. P. Coen, P. K. M. McGeehin and C. C. R. Allen, Org. Biomol. Chem., 2007, 5, 514 RSC.
- J. K. Gawronski, M. Kwit, D. R. Boyd, N. D. Sharma, J. F. Malone and A. F. Drake, J. Am. Chem. Soc., 2005, 127, 4308 CrossRef CAS.
- E. L. Eliel, Stereochemistry of Carbon Compounds, McGraw-Hill Book Company, New York, San Franciso, Toronto, London, 1962 Search PubMed; D. R. Boyd, N. D. Sharma, M. V. Berberian, K. S. Dunne, C. Hardacre, M. Kaik, B. Kelly, J. F. Malone, S. T. McGregor and P. J. Stevenson, Adv. Synth. Catal., 2010, 352, 855 Search PubMed.
- Y. Ichikawa, Synlett, 2007, 2927 CrossRef CAS; Y. Ichikawa, T. Yamaoka, K. Nakano and H. Kotsuki, Org. Lett., 2007, 9, 2989 CrossRef CAS; Y. Ichikawa, H. Egawa, T. Ito, M. Isobe, K. Nakano and H. Kotsuki, Org. Lett., 2006, 8, 5737 Search PubMed; Y. Ichikawa, M. Osada, I. I. Ohtani and M. Isobe, J. Chem. Soc., Perkin Trans. 1, 1997, 1449 RSC; Y. Ichikawa, K. Tsuboi and M. Isobe, J. Chem. Soc., Perkin Trans. 1, 1994, 2791 RSC.
- C. Stock and R. Bruckner, Synlett, 2010, 2429 Search PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- A. Palmgren, A. L. E. Larsson, J. E. Backvall and P. Helquist, J. Org. Chem., 1999, 64, 836 CrossRef CAS.
- D. R. Boyd, N. D. Sharma, B. Byrne, M. V. Hand, J. F. Malone, G. N. Sheldrake, J. Blacker and H. Dalton, J. Chem. Soc., Perkin Trans. 1, 1998, 1935 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra. See DOI: 10.1039/c2ob06904k |
| This journal is © The Royal Society of Chemistry 2012 |