Characterization of DcsC, a PLP-independent racemase involved in the biosynthesis of D-cycloserine†
David
Dietrich
,
Marco J.
van Belkum
and
John C.
Vederas
*
Department of Chemistry, University of Alberta, Edmonton, AB, Canada T6G 2G2. E-mail: john.vederas@ualberta.ca; Fax: +01-780-492-2134; Tel: +01-780-492-5475
First published on 6th February 2012
Abstract
The biosynthetic gene cluster responsible for the generation of the antibiotic D-cycloserine (DCS) has recently been disclosed. One of the putative enzymes described was DcsC, which showed a high degree of homology to diaminopimelate epimerase (DapF). Based on this homology, the activity of DcsC was presumed to be the racemization of O-ureido-L-serine, a proposed intermediate in DCS biosynthesis. Here we describe the cloning, overexpression and characterization of this enzyme. Using synthetic standards we show that DcsC is a racemase that operates on both O-ureido-L- and D-serine, and that it employs a two-base mechanism, with a thiolate–thiol pair in the active site. The activity of this enzyme was shown to be optimal at pH ∼ 7.8, with a similar kcat/KM ratio in both the L → D direction and D → L direction. Activity was abolished with thiol-inactivating reagents such as iodoacetamide and Hg2+ ions. Further evidence for a thiolate in the active site was obtained through the use of an epoxide-containing substrate analogue (6), which became covalently attached to the enzyme.
Introduction
The antibiotic D-cycloserine (2, DCS) is produced by various Streptomyces species.1 It is a structural analogue of D-alanine, and is known to inhibit two critical enzymes involved in bacterial cell wall biosynthesis: alanine racemase2 and D-alanyl–D-alanine ligase.3 This drug is commonly employed as a second-line defense against Mycobacterium tuberculosis,4 when resistance to other antibiotics is observed.5 More recently, it was tested as an NMDA receptor agonist for treatment of various psychological dysfunctions.6 Although DCS has been in clinical use for decades, interest in its biosynthesis has only recently been awakened. Specifically, the gene cluster that is involved in the biosynthesis of 2 was described.7 In all, ten genes were identified in the cluster and were assigned functions based on sequence similarity analysis. Two of these genes (dcsI and dcsJ) have previously been identified,8 and are believed to be responsible for resistance to DCS in the producing organism. Of the remaining eight genes, six are proposed to be involved in DCS biosynthesis; the function of the last two genes is unknown. Based on preliminary studies, the authors suggested a putative biosynthetic pathway for 2 (Fig. 1A).7 In the first step, L-serine is acetylated in the presence of acetyl-CoA through the action of DcsE. Acetyl L-serine is then converted to O-ureido-L-serine (1-Ll), catalyzed by DcsD, in the presence of hydroxyurea. The hydroxyurea is obtained from the hydrolysis of N-hydroxy-L-arginine by the arginase homologue DcsB. The stereochemistry of 1-Ll would then be inverted by the putative racemase, DcsC. This function was proposed based on the sequence similarity (60% identity) of DcsC to the known epimerase DapF from Xanthomonas campestris. Finally, cyclization and urea hydrolysis are suggested to be catalyzed by DcsG and DcsH. To this point, the proposed functions of DcsE and DcsB have been confirmed.7 | ||
| Fig. 1 A. Putative biosynthesis of D-cycloserine. B. Activity of DapF, a metal- and PLP-independent racemase. | ||
The hypothetical racemase DcsC displays a high degree of homology to DapF (diaminopimelate epimerase). DapF catalyzes the reversible interconversion of L,L-diaminopimelic acid (3) to meso-diaminopimelic acid (4), which is involved in lysine biosynthesis in bacteria and plants (Fig. 1B).9 This enzyme belongs to a class of metal- and pyridoxal 5′-phosphate (PLP)-independent racemases that includes proline racemase,10aspartate racemase11 and glutamate racemase.12 These enzymes are capable of inducing epimerization of the α-carbon of unactivated amino acids through the use of a thiolate–thiol pair within the active site (Fig. 2). Several mechanistic studies of these enzymes have shed light on the active-site requirements for this activity. Recent crystal structures13 of DapF indicate how a thiolate (expected pKa ∼ 10) can induce the thermodynamically unfavourable deprotonation of the α-carbon of a zwitterionic amino acid (pKa ∼ 29).14 The structural data led to the proposal that the enzyme, which initially has an open conformation and surprisingly low thiol pKa's (measured to be ca. 6.1 and 7), adopts a closed geometry upon substrate binding. This structural reorganization is required to induce catalysis and provides stabilization to the transition state as follows.13a The closure of the enzyme upon the substrate expels water from the active site and thereby desolvates the thiolate that exists at physiological pH in the open form. The resulting basic thiolate is then in a hydrophobic environment and also loses stabilization of negative charge that had been provided by an α-helix induced dipole. The substrate is rigidly held in the active site in a conformation ideal for deprotonation of the α-carbon. In particular, the dispersal of negative charge on the α-carboxylate oxygens occurs through full hydrogen bonding to protein backbone amide hydrogens and is enhanced by an α-helix dipole. Maximal stereoelectronic overlap of the newly formed carbanion with the π-system of the α-carboxylate is maintained by this rigid orientation. There is minimal motion of the substrate during catalysis. Complete protonation of the α-amine functionality enables it to remain an electron-withdrawing substituent at the α-carbon. An acidic cysteine thiol is perfectly poised on the opposite side of the α-carbon to protonate the carbanion. A key feature of such epimerases is the separation of basic and acidic sites that is induced by the structure of the protein and cannot be readily achieved in free solution.
 | ||
| Fig. 2 Mechanism of racemases that employ a thiolate–thiol pair in the active site. | ||
Aside from the conformational and dynamic requirements for catalysis, substrate selectivity of DapF is very strict. Both correct chain length and presence of all substrate amino and carboxyl groups are essential for DapF. The D,D-enantiomer of diaminopimelic acid is not a substrate for the enzyme, indicating that the stereochemistry at the non-reacting distal site is recognized by DapF.9a,15 Indeed, the crystal structures of DapF display a network of hydrogen bonds between the distal site of the substrate and the enzyme that is highly conserved in enzymes from both bacteria and higher plants (Arabidopsis thaliana).
Based on the homology that DcsC shares with DapF, we chose to test if DcsC is also a metal- and PLP-independent racemase. We describe here the cloning, purification and characterization of this putative enzyme, and demonstrate that it behaves similarly to other racemases that contain a thiolate–thiol pair in the active site. We also show that the enzyme is irreversibly inhibited when treated with a substrate analog bearing an epoxide.
Results
Expression and purification of DcsC
The DNA sequence encoding DcsC (protein accession no. BAI70377) bearing a C-terminal His6-tag, was cloned into E. coli M15 for over-expression. After cell lysis, the protein was purified using Ni2+-affinity chromatography, and pure enzyme was recovered by dialysis (∼30 mg L−1 culture). The mass of DcsC was determined through ESI-TOF mass spectrometry (31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 205 Da observed, 31205 Da expected). The protein was subjected to trypsin digestion and MS-MS sequencing, and 75% of the expected sequence was confirmed. The enzyme contains a total of six cysteine residues, which could potentially be responsible for the racemase activity.
205 Da observed, 31205 Da expected). The protein was subjected to trypsin digestion and MS-MS sequencing, and 75% of the expected sequence was confirmed. The enzyme contains a total of six cysteine residues, which could potentially be responsible for the racemase activity.
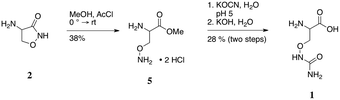
Prior to the determination of the kinetic parameters of DcsC, or in fact confirmation of the proposed activity, authentic standards of the putative substrate were needed. Specifically, the synthesis of O-ureido-D- and L-serine (1-Dd and 1-Ll) was done as shown in Scheme 1, starting from commercially available D- or L-cycloserine (2-Dd or 2-Ll).
 | ||
| Scheme 1 Synthesis of O-ureidoserine. | ||
Pure cycloserine enantiomers were individually cleaved with methanol to generate corresponding esters 5.16 These were then converted to the O-ureido species using potassium isocyanate, while maintaining the reaction at pH 5, to ensure reaction of the hydroxylamino functionality.17 Finally, saponification of the methyl esters and purification through a C-18 resin afforded the desired pure substrates 1-Dd and 1-Ll.
Kinetic analysis of DcsC
Initial studies with DcsC were conducted by 1H-NMR spectroscopy. It is expected that during the course of racemization following a two-base mechanism as shown in Fig. 2, the α-proton would exchange with the solvent.12b This was shown to be the case when DcsC was incubated in deuterated buffer (20 mM Tris–HCl, pH 8.0) containing DTT (0.1 mM) in the presence of either 1-Ll or 1-Dd. The loss of signal at δ ∼ 4.1 ppm relative to that at ∼4.3 ppm indicated exchange of the α-proton with a deuterium had occurred, while the β-methylene protons remained intact (Fig. 3). This was true with both the L- and D-substrates. These preliminary experiments indicated that the reaction was nearly 90% complete within 30 minutes. Further proof of incorporation of an α-deuteron was obtained using 2H-NMR spectroscopy (see the ESI†). The activity of DcsC (as observed by 1H-NMR) was completely abolished when the enzyme was treated with iodoacetamide prior to incubation with 1-Ll or 1-Dd. Since iodoacetamide is a potent alkylating agent, it is expected to react with any free thiols in the enzyme. This loss of activity is expected for PLP-independent racemases that contain a thiolate–thiol pair in the active site. Based on the high substrate selectivity that DapF shows, we explored whether DcsC also showed this selectivity. The same 1H-NMR assay described above was undertaken with a variety of substrates. Five amino acids were tested for deuterium incorporation in the presence of DcsC, including L-homoserine (Hse), L-Lys, L-Gln, L-Ser and L-Arg. None of these amino acids showed incorporation of deuterium at the α-position, even over a 24 hour time period (see the ESI†).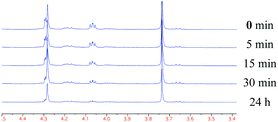 | ||
| Fig. 3 Racemization of 1-Ll by DcsC observed by 1H-NMR spectroscopy. | ||
To obtain a better idea of the kinetic parameters of DcsC, circular dichroism (CD) spectroscopy was employed. The ellipticity of 1-Ll and 1-Dd (in mdeg) showed a linear dependence with concentration at 206 nm, and thus the ellipticity could be related to concentration.18 The loss of CD signal over time in the presence of DcsC was monitored at 206 nm for both substrates 1-Ll and 1-Dd (Fig. 4A). The assays were performed at 30 °C in phosphate or borate buffer at a variety of substrate concentrations and pH values. All conditions tested contained DTT (1 mM), to prevent disulfide formation in the active site. The initial velocities of these reactions nearest the optimal pH (7.9) were used to determine the Michaelis–Menten parameters of the enzyme. In the L → D direction, the enzyme displayed a KM of 110 mM, and a kcat of 158 s−1. In the D → L direction, values of 17 mM and 29 s−1, respectively, were observed. It was somewhat surprising that the values differed so greatly based on the direction the reaction was run. When similar techniques were applied to diaminopimelate epimerase and glutamate racemase, values of kcat and KM were similar when the reaction is performed in either direction. Regardless, the catalytic efficiency of the reaction of DcsC (kcat/KM) with 1-Ll or 1-Dd is similar (1436 and 1706 s−1 M−1, respectively), as predicted by the Haldane equation. We anticipate that structural elucidation of DcsC will shed light on the discrepancy of these values.
 | ||
| Fig. 4 A. Typical changes in CD at 206 nm that were used to determine the kinetics of DcsC on substrate 1-Ll (left) and 1-Dd (right). B. Deuterium overshoots were observed by CD spectroscopy when the reaction was carried out in deuterated buffer on 1-Ll (left) and 1-Dd (right). | ||
It has been shown that enzymes that utilize a two-base thiolate–thiol mechanism for the racemization of amino acids display “deuterium overshoots” in CD spectroscopic assays.10 That is to say that when the loss of CD is observed for a protio-substrate in a deuterated buffer, a slight overshoot past a CD value of zero is observed, before the value balances to the expected zero. These “overshoots” arise from a primary kinetic isotope effect of the 2H-substrate. Since a two-base mechanism would provide a transition-state lifetime shorter than solvent exchange, each catalytic event will introduce a deuterium atom into the substrate. Since the enzyme works in both directions, an initially higher population of protio-L-substrate over the deuterio-D-substrate will generate this effect. These overshoots were observed when the CD absorbance at 206 nm of 1-Ll and 1-Dd in the presence of DcsC was monitored in deuterated buffer (Fig. 4B). This provides further evidence that DcsC operates by a similar mechanism as DapF.
Inhibition of DcsC activity
To further confirm that DcsC operates via a thiolate–thiol pair within the active site, a series of inhibition assays were done. The divalent metal-independence of DcsC was confirmed when enzyme activity was not affected by the presence of 1 mM EDTA (Fig. 5A). Introduction of 1 mM Mg2+ to the reaction also did not have an effect on the observed activity of DcsC (Fig. 5B). As seen with the 1H-NMR assay, the activity of DcsC was abolished when the enzyme was incubated with iodoacetamide prior to introduction of the substrate. These results were confirmed using a CD spectroscopic assay (Fig. 5A). DcsC was shown to retain activity in the presence of hydroxylamine (1 mM). Since hydroxylamine is an inhibitor of PLP-dependent enzymes,19 this result implies that DcsC is not a PLP-dependent enzyme. When DcsC was assayed in the presence of Hg2+ (10 μM), the activity was again completely abolished (Fig. 5B). This is the expected result for an enzyme that relies on cysteine residues for catalysis, as mercury is extremely thiophilic.9a | ||
| Fig. 5 A. Effect of non-specific inhibitors on the racemization of 1-Dd by DcsC. B. Effect of divalent cations on the racemization of 1-Dd by DcsC. C. Effect of irreversible inhibitor 6 on the racemization of 1-Dd by DcsC. | ||
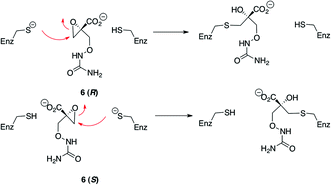
To further confirm that DcsC operates via a thiol–thiolate pair in the active site, an irreversible inhibitor was designed. Based on the proposed mechanism of DcsC, replacement of the α-proton with a suitable electrophile should generate a substrate that can form a covalent bond with the enzyme. This has previously been shown with aziridines in the case of glutamate racemase20 and diaminopimelate epimerase.21 We postulated that an epoxide could also serve this purpose, and undertook the synthesis of epoxide 6 (Scheme 2). This was synthesized as a racemate, starting from ethyl 2-hydroxymethylacrylate. Brominated substrate 7 was treated with t-butyl peroxide, using KOH as base in the presence of phase-transfer catalyst benzyltriethyl-ammonium chloride to generate mixed peroxide 8.22 Reaction of peroxide 8 with hydroxyurea and potassium t-butoxide gave 9, the ethyl ester of the desired product.23 In this reaction, the hydroxyl group of hydroxyurea adds to the substrate in a 1,4-Michael fashion, and the subsequent enolate eliminates t-butoxide. Saponification of 9 generated the desired product 6 as a racemic mixture.
 | ||
| Scheme 2 Synthesis of irreversible inhibitor 6. | ||
Initially, epoxide 6 was shown to inhibit the racemization of 1-Dd in the presence of DcsC, using 1H-NMR spectroscopy (see the ESI†). When DcsC was pre-treated with 6 for 15 minutes prior to addition of 1-Dd, no introduction of deuterium to the substrate was noted, even after 36 hours. This inhibition was also monitored using CD spectroscopy. No loss of ellipticity at 206 nm relating to substrate 1-Dd was observed in the presence of inhibitor 6. To confirm the irreversible nature of the inhibition, two conditions were used. First, the enzyme was pretreated with epoxide 6 for 20 minutes prior to introduction of the substrate. Alternatively, the substrate and inhibitor were co-injected into the enzyme solution, and the loss of CD was monitored directly. Irreversible inhibition of DcsC is marked by the fact that the extent of inhibition following a pre-treatment with epoxide 6 is much greater than inhibition following co-injection (Fig. 5C). These results imply that the inhibitor 6 indeed irreversibly inhibits the enzyme (Scheme 3). Further evidence that epoxide 6 becomes covalently attached to DcsC was obtained using mass spectrometry. Following treatment of DcsC with 6, the enzyme was subjected to ESI-TOF-MS. The m/z ratio of the enzyme treated with epoxide 6 was shown to contain a nearly equal mixture of two proteins. The difference between these two species was 176 Da, which is the expected added mass, based on the inhibitor (Fig. 6). Very small amounts of a third species were also detected. This other species was 353 Da higher than the control, implying a small amount of addition of a second inhibitor moiety. Overall, these results indicate the attachment of predominantly one inhibitor to the enzyme. Since DcsC contains six cysteine residues, yet mainly one residue was labelled with 6, this provides possible evidence for the attachment of this inhibitor at an active-site cysteine residue. Further MS-MS sequencing studies to determine the site of attachment are ongoing.
 | ||
| Fig. 6 Deconvoluted electrospray mass spectra depicting DcsC (top), and covalent attachment of epoxide inhbitor 6 (bottom). | ||
Conclusions
Recent analysis of the biosynthetic gene cluster responsible for the production of the anti-tuberculosis drug DCS provided a series of putative enzymes with predicted, but untested function. We have shown here that one of these enzymes, DcsC, displays the predicted activity, namely O-ureidoserine racemization. Through the use of synthetic standards, we were able to demonstrate racemization of 1-Dd and 1-Ll using heterologously expressed and purified DcsC in vitro. These experiments also demonstrate that this enzyme behaves similarly to metal- and PLP-independent racemases/epimerases that employ a thiolate–thiol pair in the active site. An irreversible inhibitor of DcsC based on this mechanism was successfully synthesized. Specifically, epoxide (6) proved to be an electrophilic trap for an active-site cysteine residue of the enzyme with covalent attachment observed using mass spectrometry. With strong evidence that DcsC belongs to the same group of racemases/epimerases as diaminopimelate epimerase (DapF), the three-dimensional structure of the inhibited enzyme is currently being investigated. This can also confirm the proposed concerted mechanism of DcsC, and eliminate the possibility of an elimination–addition mechanism that has been observed in serine racemases.24 Comparison of the enzymatic residues that are responsible for recognition of the non-reacting distal site of the enzyme could lead to the ability to rationally design enzymes that can induce epimerization of other amino acids.Experimental section
General
All non-aqueous reactions were performed in oven-dried glassware, under a stream of argon. Solvents were used directly, unless otherwise noted. When required, solvents were dried as follows: MeOH was distilled over CaH2; Et2O was distilled over sodium in the presence of benzophenone as indicator; DMF was dried over activated 4 Å molecular sieves; CH2Cl2 was distilled over CaH2. Silica gel was purchased from Silicycle (240–400 mesh). All NMR spectra were recorded on Varian Unity or Inova spectrometers, at the indicated frequencies at room temperature. High-resolution mass spectrometry was recorded on an Agilent 6220 oaTOF, with electrospray ionization, and MS-MS sequencing was performed on a Waters electrospray Q-TOF.Enzyme expression and purification
DNA encoding DcsC fused to a C-terminal hexahistidine tag was synthesized by BioBasic Inc. (Ontario, Canada). The dcsCcodon usage was optimized for expression in E. coli and the DNA fragment was designed with EcoRI and HindIII restriction sites adjacent to the gene in order to clone the DNA fragment into the EcoRI and HindIII restriction sites of the expression vector pQE60 (Qiagen Inc.). The resulting recombinant plasmid was introduced into E. coli M15 (pREP4) (Qiagen Inc.) and used to overexpress DcsC. E. colicells were grown in 2xYT broth at 37 °C to an A600 of 0.7 with shaking (225 rpm). The culture flasks were then put in an ice bath for 10 minutes to chill the cells and IPTG was added to a final concentration of 0.2 mM. Culture growth was continued for additional twenty hours with shaking (225 rpm) at 12 °C before cells were harvested by centrifugation (15 minutes, 8000 rpm, 4 °C). The cells were lysed using a cell disruptor (22 kpsi), in lysis buffer (0.1 M phosphate, pH 8.0, 300 mM NaCl, 1 mM EDTA, 5 mM DTT, 10 mM imidazole). The cell lysate was recovered by centrifugation (30 minutes, 15000 rpm, 4 °C), and then bound to Ni-NTA resin (Qiagen). DcsC was eluted with a gradient of imidazole (25 → 300 mM) in lysis buffer, and fractions containing the product were determined by SDS-PAGE. The enzyme was recovered by dialysis (20 mM Tris, pH 7.8, 1 mM DTT), and quantified by UV spectrocscopy (ε280 = 21345 M−1 cm−1). Average recoveries were 20–30 mg L−1 culture.
Activity monitoring by 1H-NMR
To monitor activity of DcsC, 1-Dd (12 mg, 0.06 mmol) is dissolved in deuterated Tris or phosphate buffer (600 μL, 200 mM, pD 9.0), supplemented with dithiothreitol (1 mM), at room temperature. To initiate reaction, enzyme (2 μg) is added to the reaction. The 1H-NMR spectrum is recorded at given times from addition of enzyme. To determine the incorporated deuterium, the relative area of the peak correlating to the α-CH (triplet at 4.06 ppm) is compared to the β-CH2 peak (multiplet at 4.24–4.30 ppm). The reaction was determined to be complete when the α-CH peak disappeared.Activity monitoring using CD spectroscopy
All reactions were monitored using a 1 mm quartz cell, at 30 °C, and were monitored at 206 nm. Buffered phosphate or borate was used in all cases. For overshoot experiments, buffers were prepared in deuterated water. Reactions were monitored using an OLIS globalworks CD spetrophotometer. The CD signal (in mdeg) was recorded every five seconds, for ten to twenty minutes after initiation of the reaction.A calibration curve was prepared for both 1-Dd and 1-Ll, by averaging the CD signal obtained over a five-minute period, in phosphate buffer at pH 7.9. This was repeated over a concentration range of 1–15 mM. The data were plotted in a calibration curve, and the slope was taken as the molar CD of the substrate (in mdeg mM−1).
For kinetic analysis, substrate was dissolved in buffer, containing DTT. The reaction was initiated by addition of DcsC (5 μg), and then directly placed in the spectrophotometer. This process was repeated three times for each substrate concentration and pH examined. The data were processed in Excel and GraphPad prism. The data are plotted as the average of the values recorded in the three experiments, and the error is displayed as the average of the mean. The linear range (first two minutes) was used to perform the Michaelis–Menten analyses.
General methanolysis protocol (5)
To a solution of dry MeOH (10 mL) on an ice-bath, was added acetyl chloride (0.56 mL, 7.8 mmol), and stirred under argon for 15 minutes. Cycloserine (80 mg, 0.78 mmol) was added to the solution, and the cloudy solution was warmed to room temperature for 15 minutes. A reflux condenser was attached to the flask, and the reaction was then warmed to 70 °C, and stirred for 15 hours. Insoluble material that remained at the end of the reaction was removed by filtration. To a stirring solution of the filtrate, EtOAc (100 mL) was added dropwise, with stirring, over one hour, at which time the product precipitated as a white solid.General ureidylation and saponification protocol (1)
To a stirring solution of aminoserine methyl ester 5 (64 mg, 0.31 mmol) in water (1.55 mL) was slowly added a solution of potassium isocyanate (25.2 mg, 0.31 mmol) in water (1.55 mL). Upon complete addition, the solution was determined to be pH 4–5. The pH was maintained in this range, to obtain maximal yield. When the reaction was determined to be finished by TLC (7:3, iPrOH:conc. NH4OH), the solution was adjusted to pH ∼ 1 with 1 M HCl. Solvent was removed on a rotary evaporator, and the oily product was used in the next step without purification. The desired product was prepared by saponification of crude product. The ester (1.67 mmol) was dissolved in water (16.7 mL), and a solution of KOH (2 M, 1.76 mL, 3.52 mmol) was added. The reaction was stirred at room temperature until starting material was gone by TLC (7:3, iPrOH:conc. NH4OH). The mixture was adjusted to pH ∼ 4 using 1 M HCl, and excess EtOH (10 vol.) was added to precipitate the product. The product was further purified through a Varian BondElut C18 SPE cartridge. The cartridge was washed with water (20 mL), and then the crude product was applied dissolved in water (1 mL). The product was eluted with water (20 mL), then MeCN–H2O (20 mL, 1:1). Fractions containing product were collected and lyophilized to yield the title product as a light yellow solid.
Ethyl-2-bromomethyl-acrylate (7)
A solution of ethyl-2-hydroxymethyl acrylate (260 mg, 2 mmol) in dry Et2O (20 mL) in a dry round bottomed flask was stirred at 0 °C under a stream of argon. To this was added phosphorous tribromide (0.087 mL, 0.92 mmol) dropwise via a syringe over 3 minutes. After 45 minutes, the ice bath was removed and the reaction was stirred at room temperature, until it was deemed complete by TLC (2:1, hexanes–EtOAc). Water (10 mL) and hexanes (10 mL) were added to the reaction, and the layers were separated. The aqueous phase was washed with hexanes (3 × 10 mL), all organic layers were combined, dried over Na2SO4, and solvent was removed on a rotary evaporator. The crude light yellow oil was purified by silica gel chromatography (95:5, hexanes–EtOAc) to yield a colourless liquid: 69% yield; νmax/cm−1 (neat), 2982, 2934, 2907, 1724, 1465, 1330, 1310, 1188, 1118; δH (400 MHz, CDCl3) 1.30 (t, 3H, J = 7.1 Hz, –OCH2CH3), 4.15 (d, 2H, J = 0.9 Hz, Br–CH2), 4.24 (q, 2H, J = 7.1 Hz, –OCH2CH3), 5.91 (q, 1H, J = 0.9 Hz, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 6.29 (d, 1H, J = 0.8 Hz, CH2); δC (100 MHz, CDCl3) 14.1, 29.4, 61.3, 128.9, 137.6, 164.8; ESI-HRMS (MNa)+ 214.9678 (observed), 214.9678 (expected).
CH2), 6.29 (d, 1H, J = 0.8 Hz, CH2); δC (100 MHz, CDCl3) 14.1, 29.4, 61.3, 128.9, 137.6, 164.8; ESI-HRMS (MNa)+ 214.9678 (observed), 214.9678 (expected).
Ethyl 2-t-butylperoxomethyl-acrylate (8)
To a solution of 7 (1.0 g, 5.2 mmol) in dry CH2Cl2 (50 mL), was added benzyltriethylammonium chloride (12 mg, 0.05 mmol) and t-butylhydroperoxide (0.82 mL, ∼6 M solution in decanes, 4.9 mmol), and the reaction was stirred on an ice-water bath. Crushed potassium hydroxide (0.29 g, 5.2 mmol) was added in three portions over 15 minutes, and the reaction was stirred for 90 minutes on ice, and a further 60 minutes at room temperature. The reaction was quenched with 5% citrate (30 mL), and the layers were separated. The aqueous layer was washed with CH2Cl2 (30 mL), and then, the combined organic layers were washed with water (30 mL) and brine (30 mL). The organic layer was dried (Na2SO4) and evaporated on a rotary evaporator. The crude product was purified by silica gel chromatography (95:5, hexanes–EtOAc) to yield a colourless oil. 35% yield; νmax/cm−1 (neat), 2980, 2935, 1725, 1364, 1303, 1246, 1156, 1027; δH (500 MHz, CDCl3) 1.23 (s, 9H, –OOC(CH3)3), 1.29 (t, 3H, J = 7.1 Hz, –OCH2CH3), 4.21 (q, 2H, J = 7.1 Hz, –OCH2CH3), 4.65 (dd, 2H, J = 1.3, 0.7 Hz, tBuOO–CH2), 5.89 (d, 1H, J = 1.5 Hz, CH2), 6.34 (dt, 1H, J = 1.5, 0.73 Hz, CH2); δC (125 MHz, CDCl3) 14.3, 26.4, 60.9, 73.3, 80.6, 128.2, 136.1, 165.9; ESI-HRMS (MNa)+ 225.1094 (observed), 225.1097 (expected).
1-Carboxyethyl-1-O-ureido-methoxy oxirane (9)
Peroxide 8 (202 mg, 1 mmol) and hydroxyurea (84 mg, 1.1 mmol) were dissolved in dry DMF (10 mL), and the mixture was cooled to 0 °C on an ice-water bath. To this mixture was added solid potassium t-butoxide (16.8 mg, 0.15 mmol), and the solution turned light yellow. The reaction was stirred at 0 °C, for 1 hour, and at room temperature for 5 hours. Solvent was then removed by a rotary evaporator attached to high-vacuum. The crude yellow mixture was applied to silica gel chromatography (97:3, CH2Cl2–MeOH), to yield the product as a waxy white solid: 36% yield; νmax/cm−1 (CHCl3 cast) 3466, 3188, 2986, 1728, 1690, 1588, 1185, 1143; δH (400 MHz, CDCl3) 1.30 (t, 3H, J = 7.1 Hz, –OCH2CH3), 2.98 (d, 1H, J = 5.9 Hz, oxirane-CH2), 3.17 (d, 1H, J = 5.9 Hz, oxirane-CH2), 4.08 (d, 1H, J = 11 Hz, –CH2ONH), 4.26 (qd, 1H, J = 7.1, 2.0 Hz, –OCH2CH3), 4.37 (d, 1H, J = 11.0 Hz, –CH2ONH), 5.95–5.70 (br s, 2H, NH2), 7.57 (br s, 1H, NH); δC (100 MHz, CDCl3) 14.1, 50.3, 54.2, 62.6, 75.8, 161.4, 169.1; ESI-HRMS (MNa)+ 227.0634 (observed), 227.0638 (expected).
1-Carboxy-1-O-ureido-methoxy oxirane (6)
Ester 9 (5.5 mg, 0.023 mmol) was saponified in D2O (0.7 mL) containing LiOH (2 mg, 0.046 mmol). The solvent was removed by lyophilization once the reaction was deemed to be complete by TLC (95:5, CH2Cl2–MeOH) and 1H-NMR. Quant.; νmax/cm−1 (MeOH cast), 3325, 1670, 1618, 1429, 1024; δH (500 MHz, D2O) 2.98 (s, 2H, oxirane-CH2), 3.82 (d, 1H, J = 11.4 Hz, –CH2ONH), 4.65 (d, 1H, J = 11.4 Hz, –CH2ONH); δC (125 mHz, D2O) 49.1, 57.1, 77.5, 163.4, 174.9; ESI-HRMS (M − H)− 175.0353 (observed), 175.0360 (expected).
Acknowledgements
We would like to thank Ms Jing Zheng and Mr Bela Reiz from the University of Alberta Mass Spectrometry Laboratory for their assistance in the ESI-TOF and MS-MS analyses, and Mr Wayne Moffat of the Spectral Services Laboratory for his assistance with CD spectroscopy. We would also thank Mr. Landon Kymson for his valuable help in preparing enzyme substrates. Funding was provided by Natural Sciences and Engineering Research Council of Canada (NSERC), Alberta Innovates Health Solutions and the Canada Research Chair in Bioorganic and Medicinal Chemistry.Notes and references
- P. H. Hidy, E. B. Hodge, V. V. Young, R. L. Harned, G. A. Brewer, W. F. Phillips, W. F. Runge, H. E. Stavely, A. Pohland, H. Boaz and H. R. Sullivan, J. Am. Chem. Soc., 1955, 77, 2345 CrossRef CAS.
- M. P. Lambert and F. C. Neuhaus, J. Bacteriol., 1972, 110, 978 CAS.
- (a) J. Bruning, A. Murillo, O. Chacon, R. G. Barletta and J. C. Sacchettini, Antimicrob. Agents Chemother., 2011, 55, 291 CrossRef CAS; (b) F. C. Neuhaus and J. L. Lynch, Biochemistry, 1964, 3, 471 CrossRef CAS.
- G. Pfyffer, D. A. Bonato, A. Ebrahimzadeh, W. Gross, J. Hotaling, J. Kornblum, A. Laszlo, G. Roberts, M. Salfinger, F. Wittwer and S. Siddiqi, J. Clin. Microbiol., 1999, 37, 3179 CAS.
- S. Haydel, Pharmaceuticals, 2010, 3, 2268 CrossRef CAS.
- (a) D. C. Goff, G. Tsai, J. Levitt, E. Amico, D. Manoach, D. A. Schoenfeld, D. L. Hayden, R. McCarley and J. T. Coyle, Arch. Gen. Psychiatry, 1999, 56, 21 CrossRef CAS; (b) L. J. Herberg and I. C. Rose, Pharmacol., Biochem. Behav., 1990, 36, 735 CrossRef CAS; (c) P. D. Leeson and L. L. Iversen, J. Med. Chem., 1994, 37, 4053 CrossRef CAS.
- T. Kumagai, Y. Koyama, K. Oda, M. Noda, Y. Matoba and M. Sugiyama, Antimicrob. Agents Chemother., 2010, 54, 1132 CrossRef CAS.
- M. Noda, Y. Kawahara, A. Ichikawa, Y. Matoba, H. Matsuo, D. G. Lee, T. Kumagai and M. Sugiyama, J. Biol. Chem., 2004, 279, 46143 CrossRef CAS.
- (a) M. Antia, D. S. Hoare and E. Work, Biochem. J., 1957, 65, 448 CAS; (b) C. W. Koo and J. S. Blanchard, Biochemistry, 1999, 38, 4416 CrossRef CAS.
- G. J. Cardinale and R. H. Abeles, Biochemistry, 1968, 7, 3970 CrossRef CAS.
- T. Yamauchi, S. Y. Choi, H. Okada, M. Yohda, H. Kumagai, N. Esaki and K. Soda, J. Biol. Chem., 1992, 267, 18361 CAS.
- (a) K. A. Gallo, M. E. Tanner and J. R. Knowles, Biochemistry, 1993, 32, 3991 CrossRef CAS; (b) M. E. Tanner, K. A. Gallo and J. R. Knowles, Biochemistry, 1993, 32, 3998 CrossRef CAS; (c) M. A. Spies, J. G. Reese, D. Dodd, K. L. Pankow, S. R. Blanke and J. Baudry, J. Am. Chem. Soc., 2009, 131, 5274 CrossRef CAS.
- (a) B. Pillai, M. M. Cherney, C. M. Diaper, A. Sutherland, J. S. Blanchard, J. C. Vederas and M. N. G. James, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8668 CrossRef CAS; (b) B. Pillai, M. Cherney, C. M. Diaper, A. Sutherland, J. S. Blanchard, J. C. Vederas and M. N. G. James, Biochem. Biophys. Res. Commun., 2007, 363, 547 CrossRef CAS; (c) B. Pillai, V. A. Moorthie, M. J. van Belkum, S. L. Marcus, M. M. Cherney, C. M. Diaper, J. C. Vederas and M. N. G. James, J. Mol. Biol., 2009, 385, 580 CrossRef CAS; (d) M. Stenta, M. Calvaresi, P. Altoe, D. Spinelli, M. Garavelli, R. Galezzi and A. Bottoni, J. Chem. Theory Comput., 2009, 5, 1915 CrossRef CAS.
- (a) J. P. Richard and T. L. Amyes, Curr. Opin. Chem. Biol., 2001, 5, 626 CrossRef CAS; (b) J. P. Richard and T. L. Amyes, Bioorg. Chem., 2004, 32, 354 CrossRef CAS.
- P. J. White, B. Lejeune and E. Work, Biochem. J., 1969, 113, 589 CAS.
- C. Stammer, J. Org. Chem., 1962, 27, 2957 CrossRef CAS.
- G. Zvilichovsky, Tetrahedron, 1966, 22, 1445 CrossRef CAS.
- H. Teves, T. Waniek, M. Pietzsch, C. Syldatk and M. Reuss, Fresenius J. Anal. Chem., 1999, 363, 738 CrossRef CAS.
- J. S. Wiseman and J. S. Nichols, J. Biol. Chem., 1984, 259, 8907 CAS.
- M. E. Tanner and S. C. Miao, Tetrahedron Lett., 1994, 35, 4073 CrossRef CAS.
- C. M. Diaper, A. Sutherland, B. Pillai, M. N. G. James, P. Semchuk, J. S. Blanchard and J. C. Vederas, Org. Biomol. Chem., 2005, 3, 4402 CAS.
- C. Navarro, M. Degueil-Castaing, D. Colombani and B. Maillard, Synth. Commun., 1993, 23, 1025 CrossRef CAS.
- (a) C. Cramail, M. Degueil-Castaing and B. Maillard, J. Org. Chem., 2001, 66, 3492 CrossRef CAS; (b) C. Cramay, R. Ferdinando, M. Degueil-Castaing and B. Maillard, Chem. Commun., 1998, 1855 RSC.
- V. N. Foltyn, I. Bendikov, J. De Miranda, R. Panizzutti, E. Dumin, M. Shleper, P. Li, M. D. Toney, E. Kartvelishvily and H. Wolosker, J. Biol. Chem., 2005, 280, 1754 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Including MS-MS sequencing data, and 1H and 13C spectra. See DOI: 10.1039/c2ob06864h |
| This journal is © The Royal Society of Chemistry 2012 |