 Open Access Article
Open Access ArticleDesign, synthesis, and application of tartaric acid derived N-spiro quaternary ammonium salts as chiral phase-transfer catalysts†‡
Mario Waser*, Katharina Gratzer, Richard Herchl and Norbert Müller
Institute of Organic Chemistry, Johannes Kepler University Linz, Altenbergerstraße 69, 4040 Linz, Austria. E-mail: Mario.waser@jku.at; Fax: +43 732 2468 8747; Tel: +43 732 2468 8748
First published on 17th October 2011
Abstract
A novel class of tartaric acid-derived N-spiro quaternary ammonium salts was synthesised starting from known TADDOLs. These compounds were found to catalyse the asymmetric α-alkylation of glycine Schiff bases with high enantioselectivities and in good yields.
The use of chiral phase-transfer catalysts (PTCs) for asymmetric reactions has attracted considerable interest over the last three decades.1 Among the different catalytically active structural motives like crown ethers, ammonium salts, and phosphonium salts, chiral quaternary ammonium salts have found the most widespread applications.1 Following the seminal reports of Wynberg2 and a group of Merck scientists3 describing the successful use of chiral quaternary ammonium salts for asymmetric epoxide formation2 or methylation of a phenylindanone derivative,3 cinchona-based PTCs have been thoroughly investigated in the 1990s.4–6 Due to their high catalytic potential and broad application scope, these catalysts still belong to the most commonly employed and most powerful PTCs as shown in several recent reports.7,8
In 1999, Maruoka et al. introduced C2-symmetric binaphthyl-based spiro ammonium salts as chiral PTCs.9 These Maruoka catalysts proved to be very efficient even at low catalyst loadings (<1 mol%) for a variety of asymmetric transformations.1,9,10 In addition, also Shibasaki's tartaric acid-derived bidentate PTCs11 and Lygo's spirocyclic catalysts12 were found to be highly efficient in asymmetric catalysis.
However, considering the fact that asymmetric phase-transfer catalysis using quaternary ammonium salts has been investigated for three decades now, it is somewhat surprising that besides the already mentioned privileged catalyst structures only a few other, (most of them significantly less powerful) ammonium salt-based PTCs have been reported so far.13,14
One of the main demands for novel catalysts is easy accessibility from sufficiently available chiral starting materials. Tartaric acid (1) has obtained a prominent position in asymmetric catalysis as both enantiomers are readily available in ample quantities. Although Shibasaki et al. have demonstrated the high potential of tartaric acid-derived bidentate PTCs,11 others were less successful in their attempts to synthesize powerful tartaric acid-derived catalytically active quaternary ammonium salts.13
Considering the high potential of tartaric acid-derived TADDOLs (2) as chiral ligands in (transition-) metal catalysis,15 we targeted the use of TADDOLs as starting materials for the synthesis of a new class of quaternary ammonium salts. Inspired by Maruoka's success using axially-chiral C2-symmetric binaphthyl based N-spiro quaternary ammonium ions,1,9,10 and keeping in mind that TADDOLs represent a unique moiety with electronic and steric properties different from other chiral motives, we inferred that the centro-chiral C2-symmetric N-spiro ammonium compounds 3 would be interesting targets with respect to the development of novel chiral PTCs (Scheme 1).
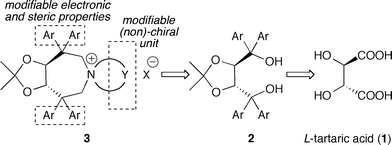 | ||
| Scheme 1 Targeted tartaric acid-based C2-symmetric PTCs. | ||
Catalyst syntheses
The syntheses of TADDOL derivatives were carefully investigated and described in the past, especially by the Seebach group.15,16 While different aryl substituents have to be introduced early in the synthesis from 1, modifications of the hydroxyl groups are most commonly achieved via conversion of 2 to dichloro compounds 4, which can then be reacted with different nucleophiles.15–17Initial experiments to develop a suitable synthesis route were carried out using the dichloride 4a (Ar = Ph)16a as the starting material. Subsequent carbon-chain elongation could be achieved using TMSCN (2.5 eq.) in the presence of catalytic amounts of SnCl4 (25%).18 With the dinitrile 5a in hand, conversion into the sec-amine 6a was thought to be just a matter of standard functional group manipulations. However, neither attempts to saponify 6a, nor conversion into the corresponding dialdehyde upon treatment with DIBAL-H were feasible due to the low reactivity of 5a (under harsher conditions partial decomposition but no product formation was observed). Using LiAlH4 at room temperature in different ethereal solvents also did not affect the cyano groups, whereas a partial decomposition was observed at higher temperatures. In addition, using other hydride donors (combined with different additives) did not give any products either.
Surprisingly, upon refluxing 5a with an excess of LiAlH4 (10 eq.) in toluene for 2 h, full consumption of 5a was observed. Besides mainly unidentified decomposition products also 10% of the sec-amine 6a could be isolated. After careful optimization of the reaction conditions, it was found that refluxing a mixture of 5a and 20 eq. LiAlH4 in mesitylene for 30–45 min gave access to 6a in 37% isolated yield. Although only modest in yield, the reaction was found to be scalable to several grams, giving the key-intermediate 6a in sufficient quantities for further elaborations. The final quaternarization could be achieved upon refluxing with 1,4-dibromobutane (CH3CN, K2CO3).
As depicted in Scheme 2, a variety of different aryl substituents was introduced successfully. Noteworthy, whereas TADDOLs with aryl groups having electronic properties similar to a phenyl group were easily converted into 6, more electron-rich as well as more electron-poor aryl group containing ones failed as the corresponding dichlorides 4 could not be obtained. The different amines 6 showed similar quaternarization behaviour as 6a, giving access to a small set of different quaternary ammonium bromides 3 in reasonable yields.19
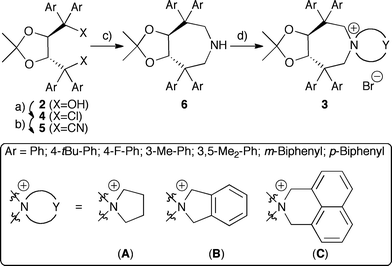 | ||
| Scheme 2 Syntheses of azepane-based PTCs 3. a) SOCl2, Et3N, CH2Cl2, 25 °C b) TMSCN, SnCl4, 25 °C, 47–63% (over two steps) c) LiAlH4, mesitylene, reflux, 29–43% d) Br-Y-Br, K2CO3, CH3CN, reflux, 49–69%. | ||
Application in asymmetric phase-transfer catalysis
Having developed a reliable route for the syntheses of C2-symmetric N-spiro ammonium salts 3, we next evaluated the catalytic potential of these compounds in the asymmetric α-alkylation of the glycine ester benzophenone Schiff base 7 with benzylbromide (8).Using the L-1 derived ammonium bromides 3aa–3ga the best-suited aryl substituent was identified first (Table 1, entries 1–8). All reactions were carried out for 20 h under biphasic conditions using 10 mol% catalyst, 25 eq. KOH, and similar dilution. As expected, the choice of the aryl group was crucial not only with respect to enantioselectivity, but also with respect to the reaction rate. Having identified the p-biphenyl containing 3ga as the best among the tested catalysts (entry 8), we next investigated the ammonium bromides 3gb and 3gc (entries 9 and 10). Whereas the o-xylene-derived catalyst 3gb gave a reduced enantiomeric excess of 51%, the naphthyl-based 3gc was found to favour the oppositely configured (R)-9, albeit in low yield and low ee only.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Cat. | Ar | Ya | Solvent | Base | T/°C | Yieldb (Conv.)c (%) | ee (%)d (Conf.)e |
| a See Scheme 2.b Isolated yields.c Judged by 1H NMR or TLC of the crude product.d Determined by HPLC using a chiral stationary phase.e Determined by comparison of the HPLC retention time and optical rotation with literature6,9 values.f Using 1% catalyst.g Using 3 eq. 8. | ||||||||
| 1 | 3aa | Ph | Aa | PhMe | KOH (50%) | rt | 70 (∼90) | 34 (S) |
| 2 | −20 | 55 (∼70) | 45 (S) | |||||
| 3 | 3ba | 4-tBu-Ph | 30 (∼50) | 61 (S) | ||||
| 4 | 3ca | 4-F-Ph | 35 (∼50) | 34 (S) | ||||
| 5 | 3da | 3-Me-Ph | 77 (∼90) | 55 (S) | ||||
| 6 | 3ea | 3,5-Me2-Ph | 47 (∼60) | 5 (S) | ||||
| 7 | 3fa | m-Biphenyl | 45 (∼60) | 7 (S) | ||||
| 8 | 3ga | p-Biphenyl | 65 (∼80) | 79 (S) | ||||
| 9 | 3gb | Ba | 57 (∼80) | 51 (S) | ||||
| 10 | 3gc | Ca | 21 (∼30) | 25 (R) | ||||
| 11 | 3ga | p-Biphenyl | Aa | PhMe | CsOH·H2O | −70 | 54 (∼70) | 39 (S) |
| 12 | CH2Cl2 | −70 | 35 (∼50) | 38 (R) | ||||
| 13 | KOH (50%) | −20 | 53 (∼70) | 28 (R) | ||||
| 14 | THF | 71 (∼90) | 7 (S) | |||||
| 15 | PhF | 82 (>90) | 68 (S) | |||||
| 16 | benzene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PhMe (2:1) :PhMe (2:1) | 69 (∼80) | 82 (S) | |||||
| 17f | 37 (∼50) | 57 (S) | ||||||
| 18 | NaOH (50%) | 33 (∼50) | 19 (S) | |||||
| 19 | CsOH (50%) | 24 (∼40) | 65 (S) | |||||
| 20 | benzene:PhMe (1:1) | KOH (50%) | −35 | 61 (∼75) | 84 (S) | |||
| 21 | PhMe | 54 (∼70) | 87 (S) | |||||
| 22g | 81 (quant) | 87 (S) | ||||||
With the ammonium bromide 3ga as the most promising among the so far synthesized PTCs we next screened different reaction conditions (entries 11–22 give a representative overview about the most significant results obtained hereby). Interestingly, the use of CH2Cl2 gave (R)-9 predominantly (entries 12, 13), whereas no stereodifferentiation could be achieved in THF (entry 14). In addition, liquid–liquid biphasic conditions were found to be superior over liquid–solid ones (entry 8 vs. 11). Besides a strong solvent effect, the choice of the base was also found to be crucial (entries 18,19). Lowering the catalyst amount to 1 mol% resulted in a reduced ee and yield (entry 17). Finally, we found that carrying out the reaction with an excess of electrophile in the presence of 10 mol% 3ga at −35 °C presents a good compromise to obtain (S)-9 in good yield and satisfactory enantioselectivity in a reasonably short reaction time (entry 22). In addition, the catalyst could be recovered (>85%) and reused several times without negatively affecting its efficiency.
Having identified the most active catalyst and the optimum reaction conditions for the asymmetric α-alkylation of 7 with benzylbromide (8) we next carried out a short screening of different electrophiles to examine the scope of this protocol. We were glad to find that other electrophiles were tolerated well, giving the corresponding products in good yields and reasonable enantioselectivities (Table 2).
| |||||
|---|---|---|---|---|---|
| Entry | RBr | Electrophile | Product | Yielda (%) | ee (%)b (Conf.)c |
| a Isolated yieldsb Determined by HPLC using a chiral stationary phase.c Determined by comparison of the HPLC retention time and optical rotation with literature values.6,9,20 | |||||
| 1 | PhCH2Br | 8 | 9 | 81 | 87 (S) |
| 2 |  | 10 | 11 | 83 | 76 (S) |
| 3 |  | 12 | 13 | 80 | 85 (S) |
| 4 | 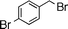 | 14 | 15 | 79 | 80 (S) |
| 5 |  | 16 | 17 | 79 | 93 (S) |
| 6 |  | 18 | 19 | 71 | 78 (S) |

In conclusion, a series of novel C2-symmetric tartaric acid-derived N-spiro quaternary ammonium salts has been successfully synthesized. The catalytic potential of these compounds in the asymmetric α-alkylation of glycine Schiff base strongly depends on their aryl substituent and on the reaction conditions. Under optimised conditions, the α-alkylated products could be obtained in good yields and up to 93% ee. Further optimizations of the catalyst structures are currently undertaken and will be reported in due course.
This work was supported by the Austrian Science Funds (FWF) Project No. P22508-N17. The NMR-spectrometers used were co-financed by the European Union in the context of the project RERI-uasb, EFRE RU2-EU-124/100-2010 (ETC Austria-Czech Republic 2007-2013).
Notes and references
- For reviews about asymmetric phase-transfer catalysis: (a) K. Maruoka, Asymmetric Phase Transfer Catalysis, WILEY-VCH, Weinheim, 2008 Search PubMed; (b) T. Shioiri, in Handbook of Phase-Transfer Catalysis, (Eds: Y. Sasson, R. Neumann), Blackie Academic & Professional, London, 1997, pp. 462–479 Search PubMed; (c) M. J. O'Donnell, in Catalytic Asymmetric Syntheses, 2nd ed. (Ed: I. Ojima), WILEY-VCH, New York, 2000, pp. 727–755 Search PubMed; (d) M. J. O'Donnell, Acc. Chem. Res., 2004, 37, 506 CrossRef CAS; (e) T. Hashimoto and K. Maruoka, Chem. Rev., 2007, 107, 5656 CrossRef CAS.
- R. Helder, J. C. Hummelen, R. W. P. M. Laane, J. S. Wiering and H. Wynberg, Tetrahedron Lett., 1976, 17, 1831 CrossRef.
- U. H. Dolling, P. Davis and E. J. J. Grabowski, J. Am. Chem. Soc., 1984, 106, 446 CrossRef CAS.
- (a) M. J. O'Donnell, W. D. Bennett and S. Wu, J. Am. Chem. Soc., 1989, 111, 2353 CrossRef CAS; (b) M. J. O'Donnell, S. Wu and J. C. Huffman, Tetrahedron, 1994, 50, 4507 CrossRef CAS.
- (a) B. Lygo and P. G. Wainwright, Tetrahedron Lett., 1997, 38, 8595 CrossRef CAS; (b) B. Lygo, J. Crosby and J. A. Peterson, Tetrahedron Lett., 1999, 40, 1385 CrossRef CAS.
- E. J. Corey, F. Xu and M. C. Noe, J. Am. Chem. Soc., 1997, 119, 12414 CrossRef CAS.
- (a) Y. Liu, B. A. Provencher, K. J. Bartelson and L. Deng, Chem. Sci., 2011, 2, 1301 RSC; (b) E. E. Maciver, S. Thompson and M. D Smith, Angew. Chem., Int. Ed., 2009, 48, 9979 CrossRef CAS; (c) L. Bernardi, E. Indrigo, S. Pollicino and A. Ricci, Chem. Commun., 2012 10.1039/c0cc05777k.
- (a) T. B. Poulsen, L. Bernardi, M. Bell and K. A. Jørgensen, Angew. Chem., Int. Ed., 2006, 45, 6551 CrossRef CAS; (b) T. B. Poulsen, L. Bernardi, J. Aleman, J. Overgaard and K. A. Jørgensen, J. Am. Chem. Soc., 2007, 129, 441 CrossRef CAS.
- T. Ooi, M. Kameda and K. Maruoka, J. Am. Chem. Soc., 1999, 121, 6519 CrossRef CAS.
- (a) S. Shirakawa, K. Liu, H. Ito and K. Maruoka, Chem. Commun., 2011, 47, 1515 RSC; (b) T. Kano, A. Yamamoto, S. Song and K. Maruoka, Chem. Commun., 2011, 47, 4358 RSC; (c) Q. Lan, X. Wang, S. Shirakawa and K. Maruoka, Org. Process Res. Dev., 2010, 14, 684 CrossRef CAS.
- (a) T. Shibuguchi, Y. Fukuta, Y. Akachi, A. Sekine, T. Ohshima and M. Shibasaki, Tetrahedron Lett., 2002, 43, 9539 CrossRef CAS; (b) A. Okada, T. Shibuguchi, T. Ohshima, H. Masu, K. Yamaguchi and M. Shibasaki, Angew. Chem., Int. Ed., 2005, 44, 4564 CrossRef CAS.
- B. Lygo, B. Allbutta and S. R. James, Tetrahedron Lett., 2003, 44, 5629 CrossRef CAS.
- (a) S. Arai, R. Tsuji and A. Nishida, Tetrahedron Lett., 2002, 43, 9535 CrossRef CAS; (b) W. E. Kowtoniuk, D. K. MacFarland and G. N. Grover, Tetrahedron Lett., 2005, 46, 5703 CrossRef CAS.
- (a) S. Kumar and U. Ramachandram, Tetrahedron, 2005, 61, 4141 CrossRef CAS; (b) S. E. Denmark, N. D. Gould and L. M. Wolf, J. Org. Chem., 2011, 76, 4260–4336 CrossRef CAS; (c) S. E. Denmark, N. D. Gould and L. M. Wolf, J. Org. Chem., 2011, 76, 4337 CrossRef CAS.
- (a) D. Seebach, A. B. Keck and A. Heckel, Angew. Chem., Int. Ed., 2001, 40, 92 CrossRef CAS; (b) H. Pellissier, Tetrahedron, 2008, 64, 10279 CrossRef CAS.
- (a) D. Seebach, A. K. Beck, M. Hayakawa, G. Jaeschke, F. N. M. Kühnle, I. Nageli, A. B. Pinkerton, P. B. Rheiner, R. O. Duthaler, P. M. Rothe, W. Weigand, R. Wünsch, S. Dick, R. Nesper, M. Wörle and V. Gramlich, Bull. Soc. Chim. Fr., 1997, 134, 315 CAS; (b) D. Seebach, M. Hayakawa, J. Sakaki and W. B. Schweizer, Tetrahedron, 1993, 49, 1711 CrossRef CAS.
- M. Waser, M. Haunschmidt and M. Himmelsbach, Monatsh. Chem., 2010, 141, 1347 CrossRef CAS.
- M. T. Reetz, I. Chatzhosifidis, H. Künzer and H. Müller-Starke, Tetrahedron, 1983, 39, 961 CrossRef CAS.
- A detailed discussion of our efforts to synthesise these PTCs as well as scope and limitations of the developed synthesis route will be reported in a full paper in due course.
- J. H. Lee, M. S. Yoo, J. H. Jung, S. Jew, H. Park and B. S. Jeong, Tetrahedron, 2007, 63, 7906 CrossRef CAS.
Footnotes |
| † This article is part of the joint ChemComm–Organic & Biomolecular Chemistry ‘Organocatalysis’ web themed issue. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and characterisation of new compounds. See DOI: 10.1039/c1ob06573d |
| This journal is © The Royal Society of Chemistry 2012 |